Синдром Марфана – это наследственное заболевание, передающееся по аутосомно-доминантному типу и характеризуется поражением соединительной ткани и ее компонентов.
Болезнь Марфана вызывается мутированием гена, кодирующего фибриллин -1.
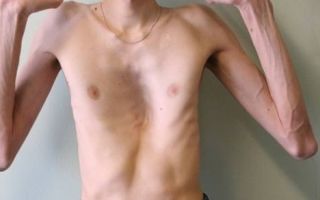
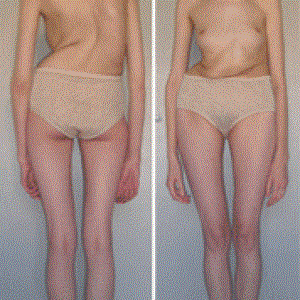
Люди с синдромом Марфана имеют удлиненные конечности, паукообразные пальцы и слабый (недоразвитый) подкожно-жировой слой и сверхгибкие суставы (см. фото ниже).
Кроме изменений костно-суставной системы, характерны изменения зрительного анализатора и сердечно-сосудистой системы. Также возможно поражение нервной, дыхательной и других систем.
Впервые описал данную патологию Вильямс, который заметил у своих брата и сестры – выпадение хрусталика, при этом они были очень высокими и имели гипермобильные суставы. Затем заметил Марфан, врач – невролог, у которого в течение 20 лет наблюдалась женщина с подобными симптомами, а затем еще 20 детей.
Причины возникновения
Болезнь Марфана у детей наследуется по аутосомно-доминантному типу (т. е передается от родителя к ребенку).
- Также возможны мутации за счет воздействия на организм женщины факторов внешней среды (ионизирующее излучение, лучевая терапия, радиация).
- Причины возникновения и механизм развития заболевания недостаточно изучены.
- Особая роль отводится нарушению процессов обмена, в результате которых накапливается в коллагеновых и эластических волокнах большое количество мукополисахаридов.
- Это ведет к тому, что соединительная ткань перерастягивается, легко подвергается механическому воздействию и приводит к развитию клинической симптоматики.
Классификация
Выделяют следующие формы болезни Марфана:
В зависимости от генной предрасположенности:
- семейная (патология передается от родителя к ребенку);
- спорадическая (патология вызвана внезапным мутированием в геноме).
В зависимости от проявлений клиники:
- стертая, когда признаки заболевания практически не проявляются и могут быть не замечены в течение всей жизни. Патологические изменения выявляются в одной или двух системах.
- выраженная, когда признаки заболевания касаются двух и более органов и систем (сердце, кости и суставы, легкие, кожа, глаза).
Симптомы болезни Марфана
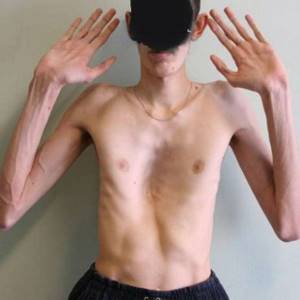
Синдромом Марфана у людей ведет к их выделению в обществе своим непропорциональным строением скелета. Для новорожденных на ранней стадии болезни, характерны длинные пальцы на руках, а к 7-9 годам у детей формируется развернутая клиническая картина.
У взрослых характерна различная симптоматика в зависимости от системы поражения:
- Нервная система: болезненность в поясничной области, головные боли,поражение симпатической и парасимпатической иннервации органов брюшной полости и малого таза (слабость кишечной стенки, недержание мочевого пузыря у ребенка). Также высок риск развития инсульта, субарахноидального кровоизлияния и разрыва аневризм головного мозга.
- Сердечно – сосудистая система: пороки сердца (сужение легочной артерии, пролабирование створок двустворчатого клапана, дилатационная кардиомиопатия, расширение границ сердца (аорты и всех ее отделов), дефекты МЖП и МЖЖ перегородок. У больных может развиваться нарушение ритма и проводимости в виде аритмий.
- Опорно-двигательный аппарат: телосложение астенической формы (дети худые), высокий рост у мужчин 190±10 см, у женщин 179±8 см, слаборазвитый подкожно-жировой слой, длинные пальцы (паукообразные), плоскостопие, череп и лицо вытянутые и узкие, недоразвитость скул, нарушение развития зубов и прикуса, вытянутая нижняя челюсть, готическое верхнее небо, гипермобильность суставов (смотрите на картинки выше). С возрастом ребенка может прогрессировать деформация позвоночного столба, с развитием сколиоза. Также может деформироваться грудная клетка, образуется вдавление – «грудь сапожника». Деформированный тазобедренный сустав нередко приводит к инвалидности при неоказанном своевременном лечении.
- Орган зрения: смещение хрусталика за счет слабого связочного аппарата) на ранней стадии, уплощение роговицы, развитие близорукости или дальнозоркости, спазм аккомодации, отслоение сетчатки.
- Кожа и мягкие ткани: перерастяжение кожи с образованием стрий атрофического характера. Они возникают внезапно, не связаны с колебание веса людей, беременностью и гормональным фоном. Кожа липкая, потная, с мраморным оттенком. Подкожно-жировой слой слабо развит, поэтому у больных наблюдаются грыжевые выпячивания в области передней брюшной стенки.
- Дыхательная система: развитие буллезной эмфиземы легких, проявляющейся кашлем, одышкой, развитием дыхательной недостаточности и спонтанного пневмоторакса.
Эндотелиальная дисфункция
Другие признаки:
Диагностика
- Диагностика основана на тщательном сборе анамнеза заболевания, выраженности клинической картины, данных осмотра, на результатах лабораторных и инструментальных методов исследований.
- Сбор анамнеза включает в себя: наличие в семье данной патологии (родители, братья, сестры) или наличие факторов, провоцирующих мутацию в геноме человека.
- К лабораторным методам относят: анализ генотипа ДНК с мутирующим геном, определение гликозаминогликанов в моче.
- К инструментальным методам исследования относят:
- ЭКГ, служит для обнаружения патологии сосудов и сердца (ССС). Выявляют характерные нарушения ритма и проводимости в виде мерцательной аритмии, желудочковой экстрасистолии, развитие дилатационной гипертрофии миокарда левого желудочка.
- ЭхоКГ, также служит для обнаружения патологии ССС. Выявляют расширение аорты и ее структур, пролабирование двустворчатого клапана, увеличение размеров левой половины сердца.
- УЗИ сердца проводится для определения осложнений (расслаивающаяся аневризма).
- Рентген органов грудной клетки (изменения скелета, расширение полостей сердца, корней легких и др.)
- Компьютерная томография, магнитно-резонансно-ядерная томография позволяет выявить патологии костно-суставной, нервной системы, нарушение кровообращения в сосудах головного и спинного мозга.
Данные методы исследования служат для обнаружения критериев синдрома Марфана в различных органах и системах. Они играют самую важную роль для постановки и подтверждения диагноза, а в последующем, для определения тактики лечения.
Существуют следующие критерии диагностики синдрома Марфана:
| Система | Большие критерии | Малые критерии |
| Опорно-двигательный аппаратДолжны быть: 4 больших критерия, либо 2 больших и 1 малый. |
|
|
| Орган зрения | Смещение хрусталика | Уплощенная роговица, близорукость, дальнозоркость, недоразвитие радужки и цилиарной мышцы глаз. |
| Сердечно-сосудистая система | Расширение аорты и ее структур | Пролабирование двустворчатого клапана, расширение клапана легочной артерии у лиц, не достигших 40 лет, отложение солей кальция на створках двустворчатого клапана, расслаивание аорты. |
| Дыхательная система | Отсутствуют | Внезапно развивающийся пневмоторакс (скопление воздуха в грудной клетке), верхушечные буллы. |
| Кожа | Отсутствуют | Повторное развитие грыжевых выпячиваний, атрофические стрии. |
| Нервная система | Расширения сосудов оболочек спинного мозга в поясничномкрестцовом отделе позвоночного столба. | Отсутствуют |
| Генетические изменения | Наличие данных критериев у родителей, детей, братьев, сестер, бабушек, дедушек. Наличие мутирующего гена, кодирующего фибриллин 1. | Отсутствуют |
Для постановки диагноза «Синдром Марфана» учитывается один признак из перечня больших критериев или малый критерий, характерный каждой из пораженной систем, кроме опорно-двигательного аппарата, (необходимо, как минимум, 4 критерия), а также наличие в семейном анамнезе больных с данной патологией.
Лечение синдрома Марфана
От синдрома Марфана полностью избавиться и устранить механизм его развития невозможно. Лечение базируется на улучшении общего состояния больного, устранении клинических проявлений и проведении профилактических мероприятий, препятствующих развитию осложнений.
Больным с данным синдромам рекомендовано ограничить физическую нагрузку до низкого уровня, или минимального. Риск появления патологии сердечно-сосудистой системы возрастает при средних и высоких физических нагрузках.
Следует обходить стороной и повседневные нагрузки, при которых возможно повышение внутригрудного давления, ведущему к развитию пневмоторакса (например, подъем тяжестей, подъем по этажам).
Больные с синдромом Марфана должны консультироваться у разных специалистов, в зависимости от клинически пораженных систем органов. Следует проходить медицинские осмотры каждые полгода в течение всей жизни.
Медикаментозное лечение
- Медикаментозная терапия направлена на устранение клинической картины заболевания.
- Со стороны сердечно-сосудистой системы рекомендованы β-адреноблокаторы (например: Анаприлин), которые снижают скорость распространения пульсовых волн при стремительно растущем расширении аорты и обратного тока крови на двустворчатом клапане или клапане аорты.
- β-адреноблокаторы также оказывают положительный эффект при нарушении ритма и проводимости, в сочетании с сердечными гликозидами.
- Но следует помнить о существующих противопоказаниях данных групп препаратов:
- Блокаторы каналов кальция используются при наличии противопоказаний к В-адреноблокаторам.
Хирургия
Хирургическое лечение проводится, если есть осложнения со стороны сердечно-сосудистой системы, с целью коррекции пораженных участков. Его проводят при пролабировании двустворчатого клапана и расслаивании аорты.
При этом осуществляется протезирование двустворчатого клапана.
У беременных с тяжелым течением болезни Марфана, роды разрешаются хирургическим путем.
Профилактика
С профилактической целью, во избежание развития инфекционных осложнений, образования тромбов и тромбоэмболии, назначаются антикоагулянты (гепарин), антибактериальная терапия и витаминотерапия.
- При синдроме Марфана с тяжелым поражением зрительного анализатора проводится хирургическая коррекция зрения, после которой пациенты должны носить очки или контактные линзы.
- Если возникают осложнения, проводят лазерную коррекцию глаукомы, катаракты, удаляют смещаемый хрусталик, заменяя его на искусственный.
- При функциональной дисфункции опорно-двигательного аппарата возникает необходимость стабилизирования позвоночника с помощью металлических пластин.
- При выраженной деформировании грудной клетки проводится торакопластика.
- При протрузии тазобедренных суставов производят внутреннее протезирование суставов.
Прогноз
Длительность жизни в среднем при синдроме Марфана составляет 30-45 лет.
Известно, что это многие знаменитые личности страдали данным синдромом. Это и Ганс Христиан Андерсен – датский писатель, автор знаменитой Русалочки; Авраам Линкольн – 16 президент США, Майкл Феллпс- известный пловец, многократный олимпийский чемпион. А также известные композиторы – Никколо Паганини, Сергей Рахманинов.
Люди с данной патологией должны тщательно следить за своим здоровьем, постоянно наблюдаться и консультироваться со своим лечащим врачом, избегать чрезмерных физических нагрузок.
По мимо медикаментозного лечения, необходимо проведение профилактических мероприятий с целью улучшения общего самочувствия, повышения иммунитета, соответствующий режим труда и отдыха.
Видеозаписи по теме
Интересное

Источник: https://tvojajbolit.ru/kardiologiya/sindrom-bolezn-marfana-chto-eto-takoe-prichinyi-vozniknoveniya-simptomyi-i-lechenie-prognoz/
Синдром Мафуччи: причины, симптомы, методы лечение, прогноз патологии
Синдром Маффуччи – это сочетанное заболевание, при котором хондродисплазия совмещается с ангиоматозом. Достаточно тяжелое состояние, которое требует длительного воздействия на больного.
При этом прогнозирование исхода терапии создать не удается из-за особенностей течения патологии.
Что такое синдром Маффуччи
- Высокую вероятность образования злокачественных опухолей;
- Высокий риск кровотечений;
- Учащение спонтанных переломов.
Синдром Маффуччи определяется в 1 случае на несколько миллионов. При этом чаще всего это врожденное состояние, спровоцированное генетическими мутациями, а чуть реже – наследственным фактором.
Синдром Маффуччи – тяжелое заболевание, которое совмещает в себе ряд проявлений. В первую очередь речь идет о хондромах, которые по сути являются доброкачественными опухолями на хрящах.
Обычно они поражают пальцы и длинные трубчатые кости. В итоге идет асимметричное искривление с последующим укорочением конечностей.
При этом нередки случаи проявления вторичного типа переломов. Данное состояние опасно тем, что доброкачественные опухоли часто переходят в злокачественные, что происходит примерно в трети случаев. Также происходит существенная деформация конечностей.
Множественные гемангиомы – вторая составляющая патологии. Это сосудистые дефекты на коже и слизистых. В некоторых случаях могут образовываться на внутренней поверхности брюшных стенок, в полости носа и так далее.
Обычно имеют синеватый оттенок и представляются в виде узелков с мягкой структурой. Повреждения этих частей могут приводить к обширным кровотечениям. По факту это сложные венозные мальформации в виде подкожных сосудов, выстланных эндотелием.
Гемангиомы имеют свойство достаточно быстро увеличиваться в размерах и поражать большие площади покровов.
Помимо того, что они имеют синеватый оттенок и мягкую структуру, эти образования также могут стать достаточно болезненными. Растут они по мере роста самого ребенка.
Они также способны влиять на форму стоп и кистей, превращая последние в достаточно уродливую массу, напоминающую виноградную гроздь.
Вместе с разрастанием хрящевых тканей указанные процессы приводят не только к существенному ограничению подвижности, но и достаточно низкому качеству и продолжительности жизни. Продолжаются негативные процессы до тех пор, пока растет и сам ребенок. Сопровождается спонтанными переломами, необратимыми формами деформации.
Причины не выявлены точно, но болезнь считают редким генетическим пороком. Соответственно и конкретных способов профилактики обнаружить не удалось. Важно отметить, что болезнь прогрессирует только пока человек продолжает расти, то есть она явно связана с процессами формирования тканей.
Симптомы
Симптоматика состояния отличается ярким проявлением буквально с первых месяцев жизни. Обычно это:
- Хондромы – хрящевые новообразования на суставах;
- Гемангиомы множественного типа – сосудистые разрастания;
- Деформация суставов;
- Болезненность кожных новообразований;
- Снижение подвижности конечностей;
- Хромота;
- Укорочение конечностей;
- Частые спонтанные переломы без причин.
При повреждении гемангиом могут проявиться обильные кровотечения венозного типа. Также стоит отметить, что деформация суставов начинается с кистей и стоп, но может распространяться и далее по конечностям и не только.
Специфической терапии на сегодня нет. Предполагается хирургическое вмешательство, которое способно устранить последствия патологии, начиная с хондром и заканчивая гемангиомами.
Но при этом чаще всего даже такое воздействие оказывается достаточно относительным, являясь только временным решением. Помимо прочего удаление сосудистых разрастаний несет риск кровотечений.
Но могут проводиться в случае с суставными деформациями и нарушениями серьезного типа такие манипуляции, как экскохлеация, остеотомия и другие. При сосудистых разрастаниях используют криодеструкцию, а также диатермокоагуляцию.
Для коррекции стоп применяют ортопедические варианты изделий, включая особую обувь и стельки.
Для того, чтобы компенсировать различную длину ног, также используется эпифизеолис, с помощью которого эти конечности удлиняются специальным аппаратом.
В целом главная угроза жизни кроется именно в высоких шансах перехода доброкачественных опухолей к злокачественным. Потому требуется периодическое обследование и удаление таких очагов.
Рентгеновский снимок таза пациента с синдромом Мафуччи
Прогноз
Прогнозы в целом относительные именно из-за неизвестных составляющих. Примерно 1/3 пациентов помимо синдрома Маффуччи получает также в анамнезе раковые опухоли. Потому без оперативного лечения обычно не обходится.
- При этом риск такого течения заболевания остается на всю жизнь, не смотря на то, что прогрессирование самого состояния останавливается вместе с ростом человека.
- Как лечат хондромы разной этиологии, смотрите в нашем видео:
Источник: http://gidmed.com/ortopediya-i-travmatologiya/sustavu-kosti/deformastya/vrozhdennye/sindrom-mafuchchi.html
Синдром Маффуччи: причины и лечение редчайшей патологии
Главная » Болезни » Опухоли » Синдром Маффуччи: причины и лечение редчайшей патологии
- 2993 0
- Синдром Маффуччи или хондродисплазия с ангиоматозом – редкое врожденное заболевание, не передающееся по наследству, несущее деформации костей, хрящей конечностей и сопровождающееся многочисленными наростами неправильной формы на поверхности кожи.
- Чаще всего новообразования в виде узелков разрастаются на конечностях, полностью видоизменяя и деформируя их.
Первый случай заболевания
Болезнь проявляется, как правило, в раннем детском возрасте и постепенно прогрессирует, принося все большие страдания. Впервые случай такого заболевания был описан в 1881 году врачом патологом А. Маффуччи. Это была женщина 40 лет с объемными наростами на руках и ногах, которые полностью лишили ее самостоятельности и возможности ходить, работать, выполнять элементарные вещи.
Деформации скелета были обнаружены с помощью рентгена, внешние проявления болезни было решено удалить. В результате операции в кровь пациентки попала инфекция, от которой та и скончалась.
Синдром Маффучи – крайне редкое встречающееся заболевание. Так, с момента его открытия в Англии было выявлено 160 случаев, в США – 100. Средняя продолжительность жизни пациентов составляет 40 лет.
Что собой представляет заболевание?
Синдром Маффуччи сопровождают:
- Хондромы – первоначально доброкачественные опухоли хрящей, чаще всего встречаются на фалангах пальцев и длинных трубчатых костях. В результате чего происходит ассиметричное искривление и укорочение конечностей, могут возникнуть вторичные переломы. Нарушается функционирование конечностей и существует большой риск перерождение в злокачественные образования (30-37%).
- Множественные гемангиомы – сосудистые образования на поверхности кожи и слизистых оболочек. Встречаются случаи наличия образований в полости носа, на языке, передней брюшной стенке и прочее. Представляют собой мягкие узелки синеватого цвета, при повреждении которых могут возникнуть обильные кровотечения.
С точки зрения строения гемангиом, это толстостенные и тонкостенные клетки кожи и подкожные сосуды, выстланные эндотелием. Их вполне можно назвать сложными венозными мальформациями.
- Так называемые, кожные сосудистые опухоли, появившись на ранней стадии заболевания, достаточно быстро увеличиваются в размере, становятся мягкими, синеватыми округлыми, иногда болезненными образованиями на коже, чаще в местах измененных костей.
- Они растут вместе с ростом ребенка, и имеют тенденцию видоизменять кисти рук и стопы в уродливые массы в виде гроздей винограда.
- Этот процесс сопровождается патологическим разрастанием хрящевой и костной тканей конечностей и метафизов длинных трубчатых костей.
- Активное развитие заболевания длится вплоть до окончания роста скелета, сопровождается спонтанными переломами и необратимыми деформациями, как внутренними, так и внешними.
Причины генетического плана
Причины проявления синдрома Маффуччи до сих пор наукой не изучены. Известно лишь, что это редкий генетический порок развития человека.
Проявляется в раннем возрасте, есть случаи, когда зачатки болезни были обнаружены у семимесячного ребенка и активно прогрессирует по мере роста человека, затем состояние стабилизируется.
Как себя проявляет аномалия?
Первые симптомы у больного ребенка проявляются чаще всего с началом ходьбы и выглядят следующим образом:
- деформация костей, хромота, укорочение конечностей;
- беспричинные переломы костей;
- образование подозрительных выростов на коже.
Также существует ряд возможных аномалий, встречающихся не всегда:
- неоднородные пятна витилиго на шее, грудной клетке, ногах и в других областях;
- атрофия одной половины мозга;
- большое количество родинок разного размера и расположения.
Клиническая картина очень сильно зависит от локализации новообразований. Часто, первые хрящевые опухоли образуются без каких-либо симптомов, проявляясь уже достигнув определенных размеров и деструктуризацией костей.
На фото наросты, характерные для синдрома маффуччи
Диагностика синдрома производится на основании общей клинической картины и результатов исследований. Как правило, назначается рентгенологическое обследование скелета человека и гистологическое исследование кожных новообразований.
Хрящевые опухоли, разрушающие и деформирующие кости, хорошо визуализируется на рентгенограмме. Они имеют характерный внешний вид и легко опознаваемы.
В случаях, когда рентген не позволяет точно поставить диагноз, назначается компьютерная или магнитно-резонансная томография, биопсия для определения природы опухолей.
Гистология новообразований
Этот анализ необходим, так как существует большой риск перехода из доброкачественных опухолей в злокачественные хондросаркомы и другие.
Подход к терапии
Несмотря на уровень развития современной медицины, медикаментозного лечения синдрома Маффуччи не существует. Применяется ортопедическое и хирургическое лечение.
Так, незначительные деформации стоп корректируют ортопедической обувью.
- Укороченность одной конечности относительно другой устраняют закрытым способом с помощью дистракционного эпифизеолиза – используя специальные медицинские аппаратов для бескровного удлинения конечностей (например аппарат Елизарова).
- При наличии окостеневших очагов разрастания хрящей показана остеотомия ̶̶ операция с рассечением кости, и дальнейшие мероприятия по ее удлинению.
- Образования на коже удаляются посредством хирургической операции – диатермокоагуляции (прижигание) или криодеструкции (разрушение с помощью холода).
- Синдром Маффуччи влияет в первую очередь на качество жизни человека, изменения в скелете и на поверхности кожи, прогрессируя, очень ограничивают больного в движениях, обслуживании себя, в работе и так далее.
Угроза жизни пациента может возникнуть при переходе клеток новообразований из доброкачественной структуры в злокачественную. Тогда применяются стандартные методы борьбы с онкологией.
Сложности и опасность
Хондродисплазия с ангиоматозом – само по себе очень сложное заболевание. Большое количество гемангиом на конечностях может привести к тому, что человек не сможет самостоятельно передвигаться и обслуживать себя.
- Являясь образованиями с большим количеством сосудов, гемангиомы могут приводить к обильным кровотечениям, в том числе и внутренним, так как были случаи удаления таких опухолей из пищевода, гортани, брюшной полости и прочее.
- Неконтролируемое разрастание хрящей чревато спонтанными переломами, не говоря уже об асимметрии тела и конечностей.
- Самым опасным для жизни является, конечно, перерождение клеток и образование саркомы.
Предупредить невозможно
Так как природа заболевания еще не изучена полностью, ясно только, что дело в генетике, способов профилактики синдрома Маффуччи не существует.
Людям, страдающим таким недугом следует своевременно обратиться к специалисту и находиться под наблюдением, чтобы контролировать процесс течения болезни.
Синдром Маффуччи – еще одна загадочная болезнь, этимология которой так и не понятна до сих пор. Генетическое врожденное заболевание проявляется в раннем возрасте и прогрессирует в течение всего периода роста скелета человека.
Хрящевая ткань разрастаясь, видоизменяет и уродует кости. Гемангиомы, в свою очередь, покрывают внешнюю поверхность тела, чаще в местах, где строение скелета уже нарушено, отрицательно влияют и на внешний вид, и на функциональные особенности.
К сожалению, полностью излечиться на сегодняшний день невозможно. В силах медицины только ортопедическим и хирургическим методами исправить деформации, облегчив пациенту существование.
Тому малому проценту населения земли, которое страдает этим заболеванием необходимо тщательно следить за его течением, своевременно делать операции и не запускать его, так как существует большой риск изменения структуры клеток новообразований в различные типы саркомы.
Источник: https://osteocure.ru/bolezni/opuholi/sindrom-maffuchchi.html
Синдром Марфана
Синдром Марфана — генетически детерминированное заболевание, характеризующееся поражением или недоразвитием соединительнотканных волокон во время эмбриогенеза и проявляющееся дисфункциональными изменениями со стороны зрительного анализатора, костно-суставной и кардиоваскулярной систем.
Синдром был открыт в 1875 году офтальмологом из Америки Э. Вильямсом.
Он обнаружил полное смещение хрусталика глаза от своего нормального положения у брата и сестры, которые имели высокий рост и чрезмерную подвижность суставов с рождения.
Спустя несколько лет детский врач из Франции А. Марфан вел наблюдения за девочкой 5 лет с прогрессирующими аномалиями скелета, чрезмерно длинными конечностями и «паучьими пальцами».
Он дал четкое описание патологии, благодаря чему синдром получил его имя.
Что это такое?
Синдром (болезнь) Марфана — аутосомно-доминантное заболевание из группы наследственных патологий соединительной ткани. Синдром вызван мутацией гена, кодирующего синтез гликопротеина фибриллина-1, и является плейотропным. Заболевание характеризуется различной пенетрантностью и экспрессивностью.
В классических случаях лица с синдромом Марфана высоки (долихостеномелия), имеют удлинённые конечности, вытянутые пальцы (арахнодактилия) и недоразвитие жировой клетчатки.
Помимо характерных изменений в органах опорно-двигательного аппарата (удлинённые трубчатые кости скелета, гипермобильность суставов), наблюдается патология в органах зрения и сердечно-сосудистой системы, что в классических вариантах составляет триаду Марфана.
Без лечения продолжительность жизни лиц с синдромом Марфана часто ограничивается 30—40 годами[2], и смерть наступает вследствие расслаивающейся аневризмы аорты или застойной сердечной недостаточности. В странах с развитым здравоохранением больные успешно лечатся и доживают до преклонного возраста.
История заболевания
В 1876 г. симптомы неизвестной патологии были отмечены доктором Вильямсом, но клинические наблюдения проводились гораздо позже — в 1896 г. педиатром из Франции А. Марфаном. Врач в течение 5-ти лет оценивал состояние девочки с неизученными ранее аномалиями, заключающимися в прогрессировании дистрофии скелета и мышечной ткани.
К середине 20-го века имелось множество описанных случаев, когда у больных наблюдались симптомы, близкие к патологии Марфана, и все они относились к заболеваниям наследственного типа.
Среди таких случаев — расслоение аорты, пороки сердца, эктопия хрусталиков, сопровождающиеся деформацией костей (грудной клетки, позвоночника) и внешними отклонениями от нормы (высокий рост, худоба, длинные конечности).
Американским генетиком МакКьюсиком было проведено детальное исследование мутаций хромосом и открыта новая группа заболеваний соединительной ткани.
Классификация
Формы патологии:
- стертая – у больных имеются незначительные изменения в 1 или 2 системах организма;
- выраженная – наличие слабовыраженных нарушений в 3 системах или характерных патологических расстройств хотя бы в 1-ой системе.
Характер течения синдрома:
- прогрессирующий – с течением времени патология нарастает и усугубляется,
- стабильный – признаки болезни на протяжении многолетних наблюдений остаются неизменными.
Этиологическая классификация:
- семейная форма — наследуется по аутосомно-доминантному принципу;
- спорадическая форма — синдром обусловлен случайной мутацией генов во время зачатия.
Причины возникновения
Это аутосомно-доминантное состояние, генетическое заболевание, передается от родителей к ребенку через гены.
Вызван мутациями в гене FBN1. Мутации FBN1 связаны с широким континуумом физических функций, от изолированных особенностей до тяжелой и быстро прогрессирующей формы у новорожденных.
Особенности расстройства чаще всего обнаруживаются в сердце, кровеносных сосудах, костях, суставах и глазах. Некоторые особенности – например, расширение аорты (расширение основного кровеносного сосуда, которое переносит кровь от сердца к остальной части тела) – может быть опасным для жизни. Также могут быть затронуты легкие, кожа, нервная система. Патология не влияет на интеллект.
Распространенность, частота встречаемости составляет 1 из 5 000 человек, включая мужчин и женщин всех рас и этнических групп.
Около 3 из 4 наследуют его, то есть получают генетическую мутацию от родителя, у которого она есть. Тип наследования при котором заболевший первый в семье- называется спонтанной мутацией.
Существует 50-процентный шанс, рождения ребенка с генетической мутации от пораженных родителей.
Люди с синдромом Марфана рождаются вместе с ним, но особенности расстройства не всегда присутствуют сразу. У некоторых людей есть много особенностей при рождении, включая серьезные состояния, такие как расширение аорты.
У других меньше симптомов, например, молодые не имеют признаков, пока не станут взрослыми. Некоторые особенности, особенно те, которые влияют на сердце и кровеносные сосуды, кости или суставы, со временем ухудшаются.
Это делает очень важным получение точной ранней диагностики и лечения. Без этого человек подвергается риску от потенциально опасных для жизни осложнений. Чем раньше началось лечение, тем лучше результаты.
Почти половина людей, страдающих синдромом Марфана, этого не знают.
Симптомы
Наиболее характерным признаком синдрома Марфана является сочетание поражений опорно-двигательной, зрительной и сердечно-сосудистой систем. Сроки их появления вариабельны, а проявления многообразны.
Обычно для постановки диагноза бывает достаточно присутствия следующих признаков:
- непропорционально длинные конечности;
- аневризма аорты;
- одно- или двухсторонняя эктопия хрусталика.
Однако кроме этих признаков существует еще около 30 других проявлений синдрома.
Поражения органов зрения
Одно- или двухсторонняя эктопия хрусталика при синдроме Марфана выявляется у 80 % пациентов. Обычно у таких людей развивается близорукость и астигматизм, но в некоторых случаях возникает и дальнозоркость. Чаще нарушения зрения происходят на 4-м году жизни ребенка. Далее они устойчиво прогрессируют.
Кроме этих заболеваний, при синдроме Марфана могут выявляться следующие патологии органов зрения:
- косоглазие;
- гипоплазия цилиарной мышцы;
- колобома радужной оболочки;
- увеличение размера и уплощение роговицы;
- изменение диаметра сосудов сетчатки;
- формирование катаракты;
- возникновение глаукомы в молодом возрасте.
Поражения скелета
У людей с синдромом Марфана могут выявляться следующие изменения в опорно-двигательной системе:
- рост намного выше среднего;
- астенический тип телосложения;
- длинный и узкий лицевой скелет;
- длинные конечности и пальцы;
- искривления позвоночного столба (спондилолистез, сколиоз, кифоз и др.);
- воронкообразная или килевидная деформация грудной клетки;
- маленький размер челюсти;
- аркоподобное высокое небо;
- чрезмерная гибкость и подвижность суставов;
- молоткообразная деформация пальцев стоп;
- протрузия вертлужной впадины;
- плоскостопие;
- патологии прикуса.
Средний рост людей с таким заболеванием при рождении может составлять у девочек 52,5 см (во взрослом возрасте около 175 см), у мальчиков 53 см (во взрослом возрасте около 191 см).
Из-за высокого неба и малых размеров челюсти у людей с синдромом Марфана могут возникать нарушения речи. Поражения скелета и суставных структур приводят к появлению артралгий и миалгий. Позднее такие изменения повышают риск развития раннего остеоартрита.
Поражения сердца и сосудов
Доминирующими и наиболее опасными проявлениями синдрома Марфана становятся признаки поражений сердца и сосудов. Появляющиеся из-за повреждения структуры стенок сосудов эластического типа изменения (особенно аорты и легочной артерии) и пороки развития клапанов, перегородок сердца вызывают следующие симптомы:
- быстрое наступление усталости;
- учащенное сердцебиение;
- стенокардические боли с локализацией в спине, верхней конечности или плече;
- холодные руки и ноги;
- аритмии;
- одышка.
При выслушивании тонов сердца у таких пациентов могут определяться шумы, а при выполнении ЭКГ выявляются признаки стенокардии. При синдроме Марфана может развиваться кистозная медиальная дегенерация митрального или аортального клапана, приводящая к пролапсу этих клапанных структур.
Кроме этого, у плода, унаследовавшего мутированные гены, с высокой долей вероятности могут развиваться врожденные пороки сердца.
Иногда, при неблагоприятном течении такой неонатальной формы синдрома, у ребенка возникает прогрессирующая сердечная недостаточность, приводящая к летальному исходу до года жизни.
Однако наиболее характерным для синдрома Марфана поражением сердечно-сосудистой системы обычно становится прогрессирующее расширение, расслоение всходящей части аорты и появление на ней аневризм.
Такие изменения провоцируются ослаблением соединительной ткани, приводящим к кистозной дегенерации сосудистой стенки. Быстрое прогрессирование таких поражений аорты может охватывать всю ее длину и ответвляющиеся от нее сосуды.
Нередко такое осложненное течение патологии приводит к смертельному исходу.
Поражения легких
Больные с синдромом Марфана не всегда имеют проблемы с легкими. Однако в некоторых случаях слабость соединительной ткани альвеол приводит к их удлинению и перерастяжению.
Впоследствии у таких больных может возникать спонтанный пневмоторакс, эмфизема легких и дыхательная недостаточность.
При отсутствии своевременной помощи и лечения такие патологии могут становиться причиной наступления летального исхода.
Кроме этого, у людей с синдромом Марфана может наблюдаться ночное апноэ, сопровождающееся прекращением дыхания во сне на 10 секунд и более.
Интеллектуальное развитие и состояние психики
У большинства детей уровень интеллекта соответствует норме и IQ составляет 85-115 единиц. У некоторых лиц с таким наследственным заболеванием уровень IQ существенно превышает верхние границы нормы.
Иногда у людей с синдромом Марфана могут присутствовать признаки неравномерной интеллектуальной деятельности и некоторые особенности личности, выражающиеся в завышенной самооценке, чрезмерной эмоциональности, плаксивости и раздражительности.
Поражения центральной нервной системы
Одним из последствий синдрома Марфана может становиться дуральная эктазия, вызывающаяся растяжением и вытяжением соединительной ткани (оболочки), обволакивающей спинной мозг. Впоследствии эта патология может приводить к появлению болей и дискомфортных ощущений в брюшной полости или к слабости и неподвижности нижних конечностей.
Поражения других систем
Кроме вышеописанных проявлений синдрома Марфана в некоторых случаях могут выявляться следующие изменения в других органах и системах:
- атрофические стрии на коже;
часто рецидивирующие бедренные и паховые грыжи; - предрасположенность к растяжению и разрывам связок, подвывихам и вывихам;
- аномальное расположение почек;
- варикозное расширение сосудов;
- опущение матки и мочевого пузыря.
Большинство детей с синдромом Марфана тяжело переносят физическую нагрузку и после нее нередко ощущают боли в мышцах. Мышцы у таких больных могут быть недоразвитыми. На фоне психоэмоционального перенапряжения у детей могут периодически возникать приступы мигренеподобной головной боли. Кроме этого, нередко ощущается слабость и признаки гипотонии.
Диагностика
Выявлением синдрома Марфана занимаются специалисты в области генетики, кардиологии, офтальмологии, неврологии, ортопедии.
Диагностика патологии включает сбор анамнеза жизни и болезни, выявление типичных клинических признаков, анализ внешнего осмотра и физикальных данных, результатов кардиографического и рентгенографического обследования, посещения офтальмолога, составление родословной у генетика.
Основные диагностические методики:
- общий анализ крови и мочи — типичные признаки воспаления;
- биохимическое исследование крови позволяет выявить дисфункцию определенного органа, возникшую в результате развития патологического процесса, а также определить первопричину заболевания и назначить правильное лечение;
- электрокардиография отображает электрические потенциалы, сформированные в работающем сердце;
- эхокардиография – исследование морфологических и функциональных изменений сердца и его клапанного аппарата;
- рентгенографическое и томографическое исследование — информативные диагностические методики, обнаруживающие поражение костей, суставов, внутренних органов и мягких тканей;
- аортография – рентгенологического исследования аорты с использованием контрастного вещества;
- УЗИ внутренних органов,
- биомикроскопия и офтальмоскопия,
- молекулярно-генетический анализ.
Как лечить синдром Марфана
Лечение синдрома Марфана всегда требует одновременного участия нескольких специалистов: кардиолога, кардиохирурга, офтальмолога, ортопеда-травматолога, терапевта.
Спектр лечебных процедур охватывает консервативные и оперативные способы лечения. Консервативные меры направлены на профилактику осложнений и поддержание нормального функционирования органов и систем, а оперативные предполагают коррекцию имеющихся анатомических изменений с целью предотвратить выраженное нарушение функций или даже угрозу для жизни больного.
Хирургические методы лечения:
- реконструктивные операции на аорте (при значительном, более 5 см, расширении восходящей части аорты и расслоении ее стенки);
- протезирование клапанов сердца;
- удаление измененного хрусталика с заменой его на искусственный;
- пластика позвоночника при выраженном сколиозе;
- эндопротезирование тазобедренных суставов;
- пластика грудной клетки (в последние годы отрицается ее целесообразность).
Больному необходим подбор очков или контактных линз, иногда возможна лазерная коррекция зрения.
Медикаментозное лечение имеет патогенетическую и симптоматическую направленность.
Больному с метаболической целью назначают большие дозы витамина С (1-3 г в сутки), препараты с глюкозаминосульфатами и хондроитинсульфатами (Терафлекс, Структум, Хондроксид, Глюкозамин, Аминоартрин, Эльбона, Юниум), Карнитина хлорид 20% раствор, Янтарную кислоту по 100-200 мг 2 раза в день, препараты магния (например, Магне В6), поливитаминно-минеральные комплексы (с кальцием, магнием, цинком, медью). Прием этих веществ направлен на нормализацию обмена веществ, укрепление соединительной ткани.
Для лечения сердечно-сосудистых нарушений часто используют β-адреноблокаторы (Пропранолол, Обзидан, Атенолол), блокаторы кальция (Нифедипин, Амлодипин, Лекоптин), ингибиторы ангиотензинпревращающего фермента (Лизиноприл, Периндоприл, Эналаприл), антиаритмические препараты (при нарушении ритма сердца). При развитии инфекционного эндокардита показаны антибиотики. После оперативных вмешательств может понадобиться антикоагулянтная терапия для снижения свертываемости крови (Фраксипарин, Клексан).
В целом подбор метода лечения и ассортимент применяемых лекарственных средств очень индивидуальны. Все зависит от спектра клинических симптомов и выраженности нарушений у конкретного больного.
Больным с синдромом Марфана показана лечебная физкультура, но в строго дозированном количестве, чтобы занятия приносили пользу сердечно-сосудистой и опорно-двигательной системам, а не вред. Нельзя заниматься контактными и игровыми видами спорта (баскетбол, футбол), рекомендовано плавание.
Прогноз
Прогноз жизни больных с синдромом Марфана определяется, в первую очередь, степенью сердечно-сосудистых изменений, а также поражений скелета и глаз.
Имеется высокий риск осложненного течения, снижения продолжительности жизни (90-95% не доживают до 40-50 лет) и внезапной смерти.
Своевременная кардиохирургическая коррекция при синдроме Марфана позволяет значительно увеличить продолжительность (до 60-70 лет) и улучшить качество жизни больных.
Больные синдромом Марфана должны находиться под постоянным врачебным наблюдением и регулярно проходить диагностическое обследование.
При синдроме Марфана показан низкий или средний уровень физической активности, исключающий занятия контактными видами спорта, спортивные соревнования, изометрические нагрузки, подводное плавание.
Женщинам детородного возраста с синдромом Марфана необходимо пройти медико-генетическое консультирование.
Источник: https://doctor-365.net/sindrom-marfana/
3Понятие о симеотике. Диагноз болезни, виды диагнозов, методологические приемы для его постановки. Прогноз заболевания
Семиотика
или семиология
(semeoticon — знак, признак), учение о симптомах
(symptom — в переводе с греческого языка
обозначает случай, признак), признаках
болезни, их происхождении, механизме
возникновения, диагностическом значении.
Симптомы,
явленные при расспросе больного,
рассматриваются как субъективные, при
объективном и дополнительном исследовании
— как объективные. Однако следует помнить,
что это деление условно, так как
субъективные симптомы иногда довольно
точно отражают сущность болезни, в то
время как объективные могут вводить
врача в заблуждение вследствие их
субъективной оценки.
Данный
раздел включает также и понятие о
синдроме. Синдром — это устойчивая
совокупность симптомов, имеющих единое
происхождение, то есть единый патогенез.
В переводе с греческого языка синдром
(syndrom) — это стечение, скопление,
совокупность.
Общая
методология и методика диагноза — учение
о диагнозе и методике его построения.
Учение
о методах распознавания болезней носит
название «диагностика».
Диагноз
(греч.)
— распознавание, различение болезней.
Понятие
«диагноз» применяется в двух смыслах:
1) название болезни; 2) мыслительный
процесс распознавания и разграничения
болезни.
Диагностический
процесс начинается с выявления признаков
или симптомов болезни. Для этого
проводится обследование больного,
которое слагается из двух основных
разделов:
- расспроса больного; '
- объективного исследования.
Объективное
исследование, в свою очередь, разделяется
на физикальное и лабораторно-инструментальное
исследования.
Физикальное
исследование (производимое врачом с
помощью его органов чувств) включает:
осмотр, ощупывание (пальпация), выстукивание
(перкуссия), выслушивание (аускультация)
и измерение.
Лабораторно-инструментальное
исследование включает: анализы крови,
мочи, других биологических сред организма,
ЭКГ, рентгенологическое, ультразвуковое,
эндоскопическое, цитологическое и др.
исследования.
Расспрос
и объективное исследование являются
основными
клиническими методами,
лабораторно-инструментальные —
дополнительными.
Правильный диагноз по данным тщательно
проведенного опроса и физикальных
методов исследования ставится у 80- 85%
больных. И лишь у 15-20% больных для
постановки диагноза требуется углубленное
исследование дополнительными методами.
4Общий осмотр: состояние больного, сознание (виды его нарушения), положение больного (виды)
Осмотр
(inspectio)
— это метод диагностического обследования
больного, основанный на зрительном
восприятии врача.
Осмотр
лучше проводить при дневном или при
рассеянном искусственном, освещении.Различают
общий
и местный осмотр.
При
осмотре оцениваются следующие показатели:
| 1) | общее состояние больного, |
| 2) | состояние сознания, |
| 3) | положение, |
| 4) | телосложение, конституция, |
| 5) | температура тела, |
| 6) | осанка, походка, |
| 7) | выражение лица, |
| 8) | голова, шея, |
| 9) | состояние кожи и видимых слизистых, |
| 10) | состояние ногтей, волосяного покрова |
| И) | подкожная жировая клетчатка, |
| 12) | лимфатические узлы, |
| 13) | состояние мышц, |
| 14) | кости, |
| 15) | суставы, |
| 16) | половое развитие. |
состояния:удовлетворительное
— при этом состоянии больной активен,
- средней тяжести — больной ограничивает движения
- тяжелое — когда больной не ходит
- крайне тяжелое состояние — положение больного пассивное, (сопор кома).
- клиническая смерть – хана,но еще жив
Сознание
- ясное сознание — больной активно ведет беседу с врачом, ориентирован в пространстве и времени;
- эйфория — представляет состояние возбуждения, неадекватное общему состоянию больного.
- ступор — плохо ориентируется в обстановке, медленно и неосмысленно отвечает на вопросы.
- сопор — спячка. Больной просыпается при сильных раздражениях,засыпает при их прекращении.
- энцефалопатей-Сопор в сочетании со спутанностью сознания
- кома — характеризуется бессознательным состоянием с отсутствием реакции на раздражители,
- Смерть мозга — это состояние, при котором мозговой кровоток отсутствует и развивается некроз всего головного мозга. При этом сохраняется сердечная деятельность, а дыхание поддерживается за счет ИВ Л. При смерти мозга на электроэнцефалограмме (ЭЭГ) регистрируется изоэлектрическая линия. ^
Положение
больного
- активное — больной меняет положение в зависимости от собственного желания;
- пассивное — лежит неподвижно, ноги и руки расслаблены, без посторонней помощи переместить их не может (крайняя степень слабости или бессознательное состояние);
- вынужденное — такое положение, которое облегчает страдания больному.
Источник: https://studfile.net/preview/6863446/page:2/