Оптическая нейропатия Лебера – это генетическое заболевание, которое проявляется прогрессирующей атрофией зрительного нерва и потерей зрения. Патология возникает при мутации ДНК в митохондриях, которая передается детям по материнской линии. Основной симптом заболевания: резкое снижение остроты зрения от нормальной до светоощущения, которое чаще всего происходит в возрасте 18-30 лет. Для диагностики назначают офтальмоскопию, ОКТ и исследование зрительных потенциалов. Подтвердить диагноз удается с помощью молекулярно-генетического теста. Лечение нейропатии Лебера включает нейрометаболические препараты и длительную реабилитацию.
Н47.2 Атрофия зрительного нерва
Заболевание названо в честь немецкого офтальмолога Теодора Лебера, который впервые описал 15 случаев внезапной потери зрения у пациентов из четырех семей. Молекулярно-генетические основы заболевания были установлены американским биохимиком Д. Уоллесом в 1988 году.
Наследственная оптическая нейропатия Лебера (НОНЛ) встречается с частотой 1 случай на 50 тыс. населения, однако носителями мутации является каждый десятитысячный житель планеты. В России заболевание чаще встречается среди жителей Сибири.
Мужчины болеют оптической нейропатией в 5 раз чаще, чем женщины.
Оптическая нейропатия Лебера
Заболевание развивается вследствие мутаций в митохондриальной ДНК, которые приводят к нарушению энергообеспечения зрительных нервов и вызывают их гибель.
До 95% случаев связано с 3 видами генетических аномалий: 3460G>A в гене ND1, 11778GC (ND6). Мутация гена ND4 встречается наиболее часто в клинической практике.
Оптическая нейропатия Лебера может быть вызвана и другими вариантами аномалий, которые недостаточно изучены из-за их редкости.
Для мутаций митохондриальной ДНК характерна неполная пенетрантность, поэтому у одних людей с аномальным геном возникает яркая клиническая картина, а другие всю жизнь остаются бессимптомными носителями. В развитии заболевания играют роль внешние триггеры. Наиболее значимыми из них признаны:
- курение;
- воздействие производственных токсинов;
- лекарственные препараты (противотуберкулезные антибиотики, глюкокортикостероиды, интерфероны).
Риск манифестации нейропатии повышается после ЧМТ, сильного стресса, острого соматического заболевания.
При нарушении последовательности нуклеотидов в митохондриальной ДНК изменяется структура белков, которые кодируются данными генами.
Поскольку при оптической нейропатии Лебера поражаются ND-гены, кодирующие протеины комплекса I (NADH-убихинон редуктаза), патология возникает на этапе образования молекул АТФ.
Клетки нервной системы чувствительны к недостатку энергетических молекул, что и обуславливает поражение зрительного нерва.
Молекулярной основой заболевания считается снижение транспорта АТФ к дистальным участкам аксонов, из-за чего на периферии запускаются процессы апоптоза.
Нехватка АТФ наиболее заметна в тонких безмиелиновых волокнах, которые составляют зрительный нерв, поэтому при нейропатии Лебера первично страдает зрение.
Развитию патологии способствует врожденный избыток аксонов в диске зрительного нерва и особое строение решетчатой пластины.
Оптическая нейропатия Лебера
В течении оптической нейропатии Лебера выделяют 3 последовательные стадии: доклиническую, острую, хроническую (атрофическую).
Первая стадия протекает бессимптомно, однако при обследовании пациента у офтальмолога по другому поводу обнаруживают отек зрительного нерва и появление на нем телеангиэктазий.
Длительность этого этапа не регламентирована, поскольку болезнь крайне редко выявляется на доклинической стадии.
Острая стадия оптической нейропатии Лебера чаще всего возникает у молодых мужчин в возрасте от 18 до 30 лет. Пациенты жалуются на резкое снижение зрения по типу центральной скотомы.
В течение 1-1,5 месяцев человек утрачивает способность различать мелкие предметы, иногда недоступен даже счет пальцев у лица, и остается лишь светоощущение.
Патология поражает оба глаза одновременно или последовательно с интервалом 6-8 недель.
Спустя 6 месяцев болезнь переходит в хроническую стадию, когда острота зрения составляет несколько тысячных.
В этот период продолжается атрофия нерва, после завершения которой функцию глазного яблока уже невозможно вернуть. Офтальмологическая картина представлена побледнением зрительного диска.
Однако существуют случаи обратного развития симптоматики, когда спустя время пациенты частично восстанавливают зрение.
При оптической нейропатии возможны экстраокулярные проявления.
Поражение нервов может затрагивать не только зрительный тракт, поэтому у части пациентов наблюдается периферическая нейропатия, миопатия, мышечная дистония.
При манифестации болезни в раннем детском возрасте есть риск развития подострой некротизирующей энцефаломиопатии (синдрома Лея). Изредка наблюдаются перекрестные симптомы НОНЛ и MELAS-синдрома.
Основная проблема нейропатии Лебера заключается в утрате зрения, которая особенно тяжело переносится молодыми больными.
На фоне слепоты развиваются тяжелые депрессии, которые могут завершаться суицидальными попытками.
Поскольку даже частичное восстановление зрения происходит не у всех, в будущем пациенты получают инвалидность и вынуждены проходить реабилитацию для адаптации к новым условиям жизни.
У некоторых женщин нейропатия Лебера протекает по типу рассеянного склероза с чередованием периодов обострений и ремиссий. У таких пациентов потеря зрения чередуется с эпизодами неполного восстановления способности видеть.
Характерно вторичное прогрессирование неврологических симптомов в сочетании со слепотой, известное как болезнь Хардинга. Патология сопровождается очагами демиелинизации в головном мозге, которые отягощают клиническое течение.
Прогрессирующее снижение зрения – повод для всестороннего обследования пациента у врача-офтальмолога. Диагностически значимыми критериями являются: центральная скотома, отсутствие болевого синдрома, наличие подобных симптомов у ближайших родственников по материнской линии. Чтобы подтвердить оптическую нейропатию Лебера, используют следующие исследования:
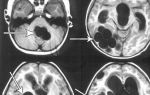
- Офтальмоскопия. При осмотре глазного дна определяется отек и восковидная бледность зрительного диска. Экскавация центральной части диска в пределах нормы. При обследовании на раннем этапе заболевания визуализируются точечные кровоизлияния в сетчатку.
- Визометрия. Оценка остроты зрения по стандартным таблицам невозможна, поскольку этот показатель не превышает 0,001. На практике это соответствует способности «счета пальцев у лица» и различения силуэтов. Дополнительно проводят периметрию, которая подтверждает выпадение центральных полей зрения.
- Оптическая когерентная томография. Прицельное исследование макулы сетчатки показывает истончение и слабую дифференцировку всех слоев, сглаживание фовеолярного контура, уплотнение внутренней пограничной мембраны. ОКТ – наиболее информативный способ диагностики атрофии ЗН.
- Исследование ЗВП. Электрофизиологическая диагностика зрительных вызванных потенциалов показывает снижение проводимости по нерву на участке до перекреста. Выраженность этих изменений коррелирует со степенью снижения зрения.
- Молекулярно-генетическое тестирование. Выделение митохондриальной мутации необходимо для верификации диагноза нейропатии Лебера. Пациентам назначают таргетное тестирование на 3 самые распространенные точечные аномалии, по показаниям проводят мультигенную панель или полное секвенирование мтДНК.
В остром периоде НОНЛ необходимо отличать от воспалительных заболеваний зрительного нерва: ретробульбарного неврита, оптикомиелита Девика, оптического неврита при ревматических заболеваниях.
Обязательно исключают ишемическую оптическую нейропатию, типичную для повышения внутриглазного давления.
Хроническую стадию болезни дифференцируют с компрессией зрительного нерва опухолями орбиты и хиазмально-селлярной области.
ОФтальмологическое обследование
В практической офтальмологии нет эффективной схемы лечения, которая могла бы улучшить зрительную функцию пациентов.
В качестве вспомогательной и патогенетической терапии назначаются препараты коэнзима Q10, левокарнитина и цитохрома. Зачастую их комбинируют в «митохондриальные коктейли» для усиления лечебного действия.
Однако даже длительный прием медикаментов не оказывает существенного влияния на клинические показатели.
Экспериментальное лечение
Поскольку медикаментозная терапия не позволяет восстановить зрение, ученые возлагают большие надежды на генно-инженерные технологии.
Их суть основана на изменении мутантной митохондриальной ДНК на нормальную, чтобы повысить содержание АТФ в аксонах зрительного пути и предотвратить атрофию нерва.
На сегодня такие методы находятся на стадии экспериментов, некоторые проходят первую стадию клинических испытаний.
Реабилитация
Для людей с инвалидностью по зрению первостепенную важность приобретает социально-психологическая адаптация.
Программа реабилитации предполагает обучение навыкам самостоятельного передвижения, ориентации в пространстве и самообслуживания.
Все пациенты проходят изучение шрифта Брайля, что позволяет им читать и продолжать образование. По желанию человека проводится трудовая реабилитации и обучение навыкам (резьба, музыка, скульптура).
Прогностически благоприятным считается НОНЛ с мутацией 14484T>C, при которой пациенты имеют шансы на полное восстановление зрения. Мутация 3460G>A протекает крайне неблагоприятно и быстро приводит к слепоте. Аномалия 11778G
Атрофия зрительного нерва Лебера: причины, симптомы, лечение
Резкое двустороннее снижение остроты зрения может говорить о развитии опасной патологии — оптической нейропатии Лебера. Такое заболевание чаще затрагивает молодых людей мужского пола. Рассмотрим причины патологии, симптомы и особенности терапии в статье.
Впервые случаи заболевания, при котором острота зрения стремительно снижается из-за атрофии зрительного нерва, были описаны в 1871 году Теодором Лебером.
Ученый наблюдал за развитием патологии у молодых людей из нескольких родственных семей, что указывало на наследственную причину заболевания.
Механизм наследования болезни был тщательно изучен, поэтому удалось доказать, что патология передается по материнской линии преимущественно мужчинам.
До сих пор характер заболевания недостаточно хорошо изучен для того, чтобы удалось найти эффективные методы лечения такой болезни. Сейчас лечение больных заключается в поддерживающей терапии, но периодически группы ученых представляют новые методики генной терапии для клинических исследований, что, конечно, вселяет надежду людям с подобным диагнозом.
Оптическая нейропатия Лебера: симптомы, лечение болезни
Любые нарушения зрения, стремительно прогрессирующие, должны быть тщательно изучены и комплексно продиагностированы. Оптическая нейропатия Лебера редко сопровождается недомоганием, характерным для неврологических заболеваний, при этом зрение падает быстро, иногда даже не в течение нескольких месяцев, а за две-три недели.
При такой остроте, зрение невозможно проверить по таблице Сивцева, потому что человек попросту не видит знаков, поэтому применяют аппаратную диагностику и метод «счета пальцев» на определенном расстоянии от лица пациента. Особенность заболевания в том, что зрительный нерв поражается из-за мутации гена.
Вырабатывается избыточное количество токсичных молекул кислорода, что негативно сказывается на состоянии клеток нерва и зрение снижается.
Оптическая нейропатия Лебера — симптомы и особенности болезни:
- молодой возраст пациентов — 18-35 лет;
- болезнь чаще развивается у мужчин;
- быстрая односторонняя, а затем и двусторонняя потеря зрения;
- острота зрения снижается в течение нескольких недель;
- нарушение цветового восприятия красного и зеленого цветов;
- патология может сопровождаться симптомами, присущими митохондриальным болезням (судороги, нарушение проводимости сердца).
При атрофии зрительного нерва Лебера чаще всего процесс развития болезни начинается с одного глаза, но нередки случаи, когда поражаются сразу два глаза. Центральное зрение снижается постоянно.
Это связано с постепенной смертью клеток зрительного нерва, передающего информацию об изображении в головной мозг. Иногда болезнь может сопровождаться симптомами, характерными для неврологических заболеваний. В этом случае больной человек страдает от дистонии, тремора, атаксии.
Реже в начале развития патологии человек замечает искры, пятна, яркие вспышки перед глазами.
Причины наследственной оптической нейропатии Лебера
Причины нейропатии Лебера, при которой атрофируется зрительный нерв, — генетические. Болезнь, обусловленная специфической мутацией генов митохондрий, передается женщинами своему потомству. Мужчины и женщины наследуют патологию от матери.
Почему так происходит? Огромное количество ДНК находится в ядре клеток и лишь небольшая его часть — в митохондриях. Гены ядра наследуются и от матери, и от отца, а вот гены митохондрии — только от матери.
Мужчина, унаследовавший мутацию, не передаст ее своим потомкам.
У значительной части людей, которые являются носителем мутированного гена, болезнь не станет явной. Более 85% женщин и 50% мужчин, которые являются носителем гена синдрома LHON, болезнь не затронет.
Причины, влияющие на прогрессирование заболевания, не ясны, но достоверно известно, что спровоцировать болезнь может неблагоприятная экологическая обстановка, стрессы, инфекции, токсическое воздействие табака и алкоголя.
Диагностика оптической нейропатии Лебера
Диагностика болезни осложнена схожестью ее симптомов с ишемической невропатией или с определенными патологиями — например, с рассеянным склерозом.
Сложно отследить и анамнез, так как не у всех носителей мутировавшего гена развивается нейропатия Лебера.
Расширенное офтальмологическое обследование может только установить наличие патологии, а вот для определения причины, понадобится генетическая диагностика.
Особенности диагностики наследственной оптической нейропатии Лебера:
- общее клиническое обследование;
- анализ митохондриальной ДНК;
- исследование полей зрения;
- осмотр глазного дна;
- когерентная томография и электроретинография (необходимы, чтобы исключить патологии сетчатки).
Специалистам рекомендуется проводить дифференциальную диагностику синдрома LHON, сравнивая симптоматику и данные обследования с другими заболеваниями, поражающими глазной нерв. Например, с ишемической, токсической невропатией. Полное офтальмологическое обследование показывает, что диск зрительного нерва воспаленный, также заметны телеангиэктазии сосудов (расширение мелких капилляров).
Наследственная оптическая нейропатия Лебера — можно ли вылечить болезнь?
Пока наследственная нейропатия оптическая считается неизлечимым заболеванием, но учеными ведется постоянный поиск новых путей для терапии подобной патологии. Благодаря методам генной терапии можно существенно замедлить прогрессирование болезней и улучшить качество жизни пациентов.
Стремительное прогрессирование болезни Лебера приводит к быстрой потере зрения людьми, которые всю свою жизнь видели нормально и не имели проблем со зрением.
Неутешительный диагноз, а также информация о том, что болезнь пока не лечится, серьезно сказываются на самочувствии таких пациентов, которые не были готовы к инвалидности.
Многие из существующих методик лечения патологии оказались малоэффективными, в том числе и хирургическое вмешательство.
К счастью, ученые, занимающиеся редкими патологиями, прикладывают немало усилий к тому, чтобы найти средство излечения. Особую надежду специалисты возлагают на генную терапию, и у многих исследовательских групп уже есть неплохие результаты.
Иногда препятствием к продолжению экспериментов является их этическая сторона, так как дальнейшие исследования предполагают использование разработанной методики на людях.
Есть ли успехи хотя бы с использованием экспериментальных моделей? Да, генетикам уже удалось найти путь к решению проблемы.
Так, группа ученых из Майами, используя экспериментальные модели, доказала, что мутировавшие гены можно безопасно заменить здоровыми, это предотвратит ухудшение питания клеток зрительного нерва.
Для исправления генетического дефекта в митохондрии необходимо ввести нормальную ДНК — это позволит исправить нарушение и восстановить зрительную функцию.
Ученые сообщают, что такой подход будет эффективным и в отношении других заболеваний, вызванных митохондриальными мутациями, а также различных нарушений, связанных с процессами старения организма.
Нейропатия Лебера: лечение и поддерживающая терапия
На данный момент медики могут предложить поддерживающую терапию, которая замедляет прогрессирование патологии. Пациентам необходимо кардинально пересмотреть уклад жизни, снизить уровень стресса и влияние негативных факторов на здоровье, отказаться от курения и алкоголя.
О появившейся проблеме обычно сигнализирует отек слоя нервных волокон (диагностика с использованием когерентной томографии). Также обнаружить изменения помогут цветовые тесты, которые сообщают о проблеме с распознаванием зеленого и красного цветов. Людям, у которых в семье были случаи оптической нейропатии Лебера, необходимо не забывать о высоких рисках развития этой опасной патологии.
Существуют разные методы терапии заболевания и пока ни один из них не имеет доказанной эффективности. Лечение ведется специалистами с учетом степени нарушения, а также имеющихся сопутствующих заболеваний у пациента, поэтому используются комбинированные протоколы терапии.
Поддерживающая терапия при атрофии зрительного нерва:
- стимуляция кровообращения в сохранившихся нервных волокнах;
- применение сосудорасширяющих средств, стимуляторов;
- лечение нейропротекторами (средствами, предупреждающими повреждение нейронов мозга);
- применение препаратов, улучшающих обменные процессы на клеточном уровне;
- физиотерапевтические процедуры, ультразвуковая терапия, электрофорез с лекарственными препаратами и травами, магнитотерапия, электростимуляция зрительного нерва;
- сбалансированное питание — в рацион необходимо включить продукты с высоким содержанием витаминов С, B1, B12;
- биостимуляторы (алоэ, экстракт стекловидного тела);
- комплексные витаминные препараты, глазные капли с витаминами.
Что касается прогноза заболевания, то в каждом случае он индивидуален, зачастую все зависит от возраста пациента. Чем моложе заболевший человек, тем выше шансы на то, что зрение хотя бы частично восстановится.
Полная слепота развивается в редких случаях.
При таком заболевании важно соблюдать все предписания специалиста, ограничить воздействие токсинов на организм (отказаться от вредных привычек, исключить прием токсичных лекарственных препаратов, некоторых видов антибиотиков).
Сложности в поддерживающей терапией возникают обычно на местах, ведь многие врачи не учитывают генетическую причину болезни как основную и ставят ошибочные диагнозы. Повышение уровня информированности о таком заболевании, как оптическая нейропатия Лебера, значительно увеличивает шансы на то, что на эту проблему будет обращено внимание разных специалистов и ученых генетиков.
Потеря зрения по наследству: что важно знать о врожденном амаврозе Лебера
Какие процессы отвечают за нашу способность видеть и как устроен «ген зрения»? Из-за чего возникает болезнь, при которой зрение полностью падает еще в детстве, и по каким признакам можно ее определить? Как живут люди, которым слепота передалась по наследству, и чем медицина может им помочь? Вместе с информационно-просветительским гуманитарным проектом «12 месяцев» мы продолжаем серию материалов о редких (орфанных) генетических заболеваниях и жизни людей с ними. Читайте в декабре рассказ о врожденном амаврозе (слепоте) Лебера, который встречается у одного из 33 тысяч человек, а также историю пациента — педагога и радиоведущей Цындымы Бойко.
 Какие процессы отвечают за нашу способность видеть и как устроен «ген зрения»? Из-за чего возникает болезнь, при которой зрение полностью падает еще в детстве, и по каким признакам можно ее определить? Как живут люди, которым слепота передалась по наследству, и чем медицина может им помочь? Вместе с информационно-просветительским гуманитарным проектом «12 месяцев» мы продолжаем серию материалов о редких (орфанных) генетических заболеваниях и жизни людей с ними. Читайте в декабре рассказ о врожденном амаврозе (слепоте) Лебера, который встречается у одного из 33 тысяч человек, а также историю пациента — педагога и радиоведущей Цындымы Бойко.
Какие процессы отвечают за нашу способность видеть и как устроен «ген зрения»? Из-за чего возникает болезнь, при которой зрение полностью падает еще в детстве, и по каким признакам можно ее определить? Как живут люди, которым слепота передалась по наследству, и чем медицина может им помочь? Вместе с информационно-просветительским гуманитарным проектом «12 месяцев» мы продолжаем серию материалов о редких (орфанных) генетических заболеваниях и жизни людей с ними. Читайте в декабре рассказ о врожденном амаврозе (слепоте) Лебера, который встречается у одного из 33 тысяч человек, а также историю пациента — педагога и радиоведущей Цындымы Бойко. Что такое врожденный амавроз Лебера и как проявляется это заболевание?
Врожденный амавроз Лебера — это тяжелое наследственное заболевание сетчатки, которое приводит к сильному ухудшению зрения и слепоте. Оно встречается у одного человека из 33 тысяч во всем мире. Уже на первом году жизни ребенка родители замечают, что младенец не фиксирует взгляд, а его зрачок не реагирует на свет.
Среди других симптомов — неконтролируемые ритмичные движения глазных яблок (нистагм), постоянное надавливание на глаза и трение глаз костяшками пальцев (симптом Франческетти). Полная потеря зрения, как правило, наступает к 10 годам или подростковому возрасту.
Врожденный амавроз или слепота (от греческого слова amauros — «темный, слепой») Лебера — это генетическое заболевание, то есть его причина — неправильная работа генов.
Оно составляет более 5% от всех унаследованных заболеваний сетчатки: известно, что с ним родились около 20% слепых детей, обучающихся в специализированных образовательных учреждениях. В 1869 году немецкий офтальмолог Теодор Лебер впервые выявил необычную и тяжелую форму потери зрения у ребенка.
Чуть позже, наблюдая за детьми в школе для незрячих, он заметил, что заболевание имеет «кровное родство» — так появилась его статья, в которой врач рассуждал о том, что слепота передается по наследству.
Предположения Лебера впоследствии подтвердились, а описанные им случаи атрофии зрительных нервов разошлись на два самостоятельных заболевания — с разными генетическими причинами возникновения, типом наследования, клиническими проявлениями и методами лечения. Это — наследственная оптическая нейропатия Лебера (Leber hereditary optic neuropathy, LHON) и амавроз зрительного нерва Лебера (Leber congenital amaurosis, LCA).
В 1957 году шведский психиатр Карл-Генри Альстрём предположил, что описанная Лебером слепота наследуется аутосомно-рецессивно.
То есть ген с ошибкой унаследован от обоих родителей, и половая принадлежность здесь не при чем — этот ген расположен не в половых хромосомах (не в X, и не в У). Просто мужчина и женщина оказались его носителями, причем клинические признаки заболевания у них, вероятно, отсутствовали. Риск, что ребенок такой пары родится с наследственным заболеванием, например, амаврозом Лебера, составляет 25%.
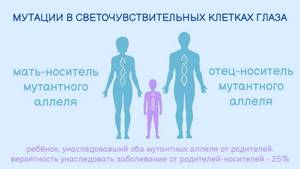
Схема аутосомно-рецессивного типа наследования врожденного амавроза Лебера
В 1992 году Олаф Рисс с коллегами подтвердили аутосомно-рецессивный характер наследования заболевания, проанализировав 23 семьи, в которых встречался амавроз Лебера. В 1995 году Аньес Камузат с коллегами обнаружили на 17 хромосоме ген, ассоциированный с развитием слепоты, — они обозначили его как LCА1.
Врожденный амавроз Лебера — это группа заболеваний?
На сегодняшний день известно как минимум 400 мутаций в 25 генах, приводящих к врожденому амаврозу Лебера. Эти генетические ошибки обнаруживаются у 70-80% пациентов — у части пациентов с клинической картиной и офтальмологическими признаками этого заболевания так и не удается найти мутацию.
Выделяют 19 типов амавроза Лебера — по типу обнаруженной мутации. Каждому типу присваиваются порядковые номера — от LCA1 до LCA19. Наиболее распространенный (5-10% всех случаев), хорошо изученный с молекулярной точки зрения и имеющий генетическую терапию — это второй тип — LCA2.
При этом типе происходит мутация в гене RPE65.
Основной механизм возникновения заболевания можно объяснить тем, что в особых светочувствительных клетках глаза — палочках и колбочках, которые преобразуют световой импульс в нервный сигнал, воспринимаемый головным мозгом, происходит нарушение зрительного цикла. При мутациях эти светочувствительные клетки погибают и перестают преобразовать сигнал, что ведет к прогрессирующей потере зрения.
Лучше всего такой патогенетический механизм изучен при мутации в гене RPE65. При этой мутации есть дефицит или полностью отсутствует фермент, который поддерживает зрительный цикл.
Без фермента разрывается цепочка последовательных химических реакций, необходимых для перевода светового импульса в «зрительную картинку», и чем меньше этого фермента, тем хуже становится зрение. Современная диагностика врожденного амавроза Лебера основана на стандартных офтальмологических методах обследования — это клинический осмотр, визометрия, авторефрактометрия. Также необходимы специальные методы — электрофизиологическое исследование, исследование полей зрения и применение оптической когеретной томографии. Также врачу необходимо собрать подробный анамнез, узнать семейную историю заболевания и провести генетическое тестирование. Зачем оно нужно? Знание точной мутации может кардинально изменить лечение амавроза Лебера, ведь это одно из немногих «редких» заболеваний, для которого разработана генная терапия. То есть неправильно работающий ген, из-за которого возникло заболевание, можно исправить. Генная терапия — это единственный метод лечения, который влияет на причину заболевания. До ее появления все лечение сводилось к поддерживающим методам — применению витаминов и препаратов, влияющих на кровоснабжение глаз, но они лишь слегка замедляли неизбежное — потерю зрения.
 Современная диагностика врожденного амавроза Лебера основана на стандартных офтальмологических методах обследования — это клинический осмотр, визометрия, авторефрактометрия. Также необходимы специальные методы — электрофизиологическое исследование, исследование полей зрения и применение оптической когеретной томографии. Также врачу необходимо собрать подробный анамнез, узнать семейную историю заболевания и провести генетическое тестирование. Зачем оно нужно? Знание точной мутации может кардинально изменить лечение амавроза Лебера, ведь это одно из немногих «редких» заболеваний, для которого разработана генная терапия. То есть неправильно работающий ген, из-за которого возникло заболевание, можно исправить. Генная терапия — это единственный метод лечения, который влияет на причину заболевания. До ее появления все лечение сводилось к поддерживающим методам — применению витаминов и препаратов, влияющих на кровоснабжение глаз, но они лишь слегка замедляли неизбежное — потерю зрения.
Современная диагностика врожденного амавроза Лебера основана на стандартных офтальмологических методах обследования — это клинический осмотр, визометрия, авторефрактометрия. Также необходимы специальные методы — электрофизиологическое исследование, исследование полей зрения и применение оптической когеретной томографии. Также врачу необходимо собрать подробный анамнез, узнать семейную историю заболевания и провести генетическое тестирование. Зачем оно нужно? Знание точной мутации может кардинально изменить лечение амавроза Лебера, ведь это одно из немногих «редких» заболеваний, для которого разработана генная терапия. То есть неправильно работающий ген, из-за которого возникло заболевание, можно исправить. Генная терапия — это единственный метод лечения, который влияет на причину заболевания. До ее появления все лечение сводилось к поддерживающим методам — применению витаминов и препаратов, влияющих на кровоснабжение глаз, но они лишь слегка замедляли неизбежное — потерю зрения. Почему же глаза идеально подходят для генной терапии? Все дело в особенностях строения и расположения глаза. Так:
- глаза — легкодоступный орган для проведения манипуляций, в отличие от, например, печени или мышц;
- исправить ген нужно в сравнительно небольшом количестве клеток;
- гемато-ретинальный барьер (особая структура, которая предотвращает проникновение лишних веществ в ткань сетчатки) позволяет ограничить распространение генной терапии вне глаза и снизить вероятность иммунного ответа, который может возникнуть из-за лечения.
Как работает первый и единственный препарат против поломки гена, ответственного за слепоту?
В 2017 году одно из самых влиятельных агентств в мире — управление по санитарному надзору за качеством пищевых продуктов и медикаментов США (Food and Drug Administration, FDA) — одобрило первый в мире препарат для лечения наследственных заболеваний сетчатки — Luxturna (Люкстурна). Он предназначен для людей с мутацией в гене RPE65 ( о нем мы говорили чуть выше).
Как работает Люкстурна? Под сетчатку каждого глаза проводится инъекция нормальной «рабочей» копии гена RPE65. Эта манипуляция позволяет доставить нужный ген непосредственно в клетки сетчатки глаза. Для такой доставки используется специальный курьер — природный адено-ассоциированный вирус, его называют вектором (Adeno-associated dependoparvovirus, AAV). Доставленный ген начинает производить фермент, необходимый для зрительного цикла.
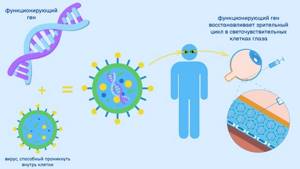
Схема генной терапии
Разработка препарата велась с 1995 года. Первые доклинические испытания проводились на собаках, которые имели ранние и тяжелые нарушения зрения и мутацию в обеих копиях гена RPE65. Эти исследования показали, что вводимая генно-инженерная конструкция не токсична, приводит к значительному улучшению зрения и останавливает гибель светочувствительных клеток. Дальше — на следующей фазе — клинические исследования проводились уже на людях. Обязательное условие для терапии — это сохранность фоторецепторов (светочувствительных клеток), так как Люкстурна не может заново «построить» новые палочки и колбочки — препарат может только поддерживать зрительный цикл в сохранившихся клетках. Есть и побочные эффекты — боль и отечность глаз, катаракта и повышение внутриглазного давления. Один из участников клинических исследований — старшеклассник Кристиан Гуардино — так описывал свое состояние после лечения: «Моя жизнь намного улучшилась», а его мать Бет Гуардино дополнила: «Теперь он может выходить на улицу в сумерки, чего не мог раньше, и он впервые увидел звезды. И он может читать мое выражение лица и знать, счастлива я или нет». Идут разработки и клинические исследования и других препаратов, которые пытаются исправить мутации не только в гене RPE65, но и остальных — например, в гене фоторецептора CEP290. В 2016 году в России появилась межрегиональная общественная организация — «Чтобы видеть». Ее цель — помочь детям и взрослым с различными наследственными заболеваниями сетчатки, приводящими к сильному ухудшению зрения вплоть до его полной потери.
 Обязательное условие для терапии — это сохранность фоторецепторов (светочувствительных клеток), так как Люкстурна не может заново «построить» новые палочки и колбочки — препарат может только поддерживать зрительный цикл в сохранившихся клетках. Есть и побочные эффекты — боль и отечность глаз, катаракта и повышение внутриглазного давления. Один из участников клинических исследований — старшеклассник Кристиан Гуардино — так описывал свое состояние после лечения: «Моя жизнь намного улучшилась», а его мать Бет Гуардино дополнила: «Теперь он может выходить на улицу в сумерки, чего не мог раньше, и он впервые увидел звезды. И он может читать мое выражение лица и знать, счастлива я или нет». Идут разработки и клинические исследования и других препаратов, которые пытаются исправить мутации не только в гене RPE65, но и остальных — например, в гене фоторецептора CEP290. В 2016 году в России появилась межрегиональная общественная организация — «Чтобы видеть». Ее цель — помочь детям и взрослым с различными наследственными заболеваниями сетчатки, приводящими к сильному ухудшению зрения вплоть до его полной потери.
Обязательное условие для терапии — это сохранность фоторецепторов (светочувствительных клеток), так как Люкстурна не может заново «построить» новые палочки и колбочки — препарат может только поддерживать зрительный цикл в сохранившихся клетках. Есть и побочные эффекты — боль и отечность глаз, катаракта и повышение внутриглазного давления. Один из участников клинических исследований — старшеклассник Кристиан Гуардино — так описывал свое состояние после лечения: «Моя жизнь намного улучшилась», а его мать Бет Гуардино дополнила: «Теперь он может выходить на улицу в сумерки, чего не мог раньше, и он впервые увидел звезды. И он может читать мое выражение лица и знать, счастлива я или нет». Идут разработки и клинические исследования и других препаратов, которые пытаются исправить мутации не только в гене RPE65, но и остальных — например, в гене фоторецептора CEP290. В 2016 году в России появилась межрегиональная общественная организация — «Чтобы видеть». Ее цель — помочь детям и взрослым с различными наследственными заболеваниями сетчатки, приводящими к сильному ухудшению зрения вплоть до его полной потери. На портале представлена информация о заболеваниях, возможностях их диагностики и терапии, устройстве быта и вспомогательных средствах, которые могут облегчить жизнь слабовидящим людям.
Анастасия Кадыкова
Врач клинической лабораторной диагностики Федерального медико-биологического агентства России
Наследственная атрофия зрительных нервов Лебера: генетика, лечение
Наследственная оптическая нейропатия Лебера (англ.: Leber optic atrophy = Leber hereditary optic neuropathy, LHON) – это редкое наследственное заболевание, вызывающее нарушения зрения. Болезнь чаще всего встречается в возрасте 27-34 лет, преимущественно поражает мужчин.
Болезнь была впервые диагностирована немецким офтальмологом Альбрехтом фон Граефом в 1858 г, но название получила в честь его ассистента Теодора Лебера, позже описавшего клиническое течение заболевания у 15 пациентов. Атрофия Лебера – первое заболевание, связанное с материнской наследственностью и специфической точечной мутацией в митохондриальной ДНК (мтДНК).
Диагностика болезни затруднена из-за низкой заболеваемости, свидетельствующей о наличии этого расстройства в семье. Для исключения других причин нарушения зрения необходимо офтальмологическое обследование. Целесообразно выполнение генетического исследования для подтверждения мутации.
Патогенез, этиология, причины
Причина болезни Лебера – генетическая мутация в ДНК, происходящая в митохондриях.
Митохондрии – это органеллы в клетках, ответственные за клеточный энергетический обмен. При этой болезни практически исключительно затрагиваются RGB, формирующие зрительный нерв.
Одно из возможных объяснений селективного поражения RGB – их высокая потребность в непрерывной доставке АТФ (аденозинтрифосфата, англ.: ATP). Гистохимические исследования показали повышенное накопление митохондрий в области пластинки cribrosa sclerae, где немиелинизированные нервные волокна выступают из сетчатки, образуя зрительный нерв.
Эта область богата ферментами Na+/K+ATP, что делает локальное нервное ведение чрезвычайно сложным процессом, и может объяснить исключительную уязвимость волокон зрительного нерва. Дефект митохондриального метаболизма приводит к локальному застою аксоплазмы с отеком. В дальнейшем это способствует дегенерации слоя RGB и их аксонов, образующих зрительный нерв.
Вопреки этой теории, фоторецепторы, сохраняющиеся при заболевании, имеют более высокие окислительные потребности, чем RGB.
Кроме того, другие митохондриальные заболевания с более тяжелым комплексным расстройством не всегда приводят к развитию атрофии зрительного нерва.
Поэтому возможно, что RGB более чувствительны к незначительным отклонениям окислительно-восстановительного потенциала клеток и образованию радикалов кислорода, чем к дефициту АТФ.
Генетика: как, кому и когда передается болезнь
Митохондриальный шаблон наследования синдрома Лебера
Наследственная атрофия зрительных нервов Лебера опосредуется мутацией ДНК в митохондриях, которую человек (преимущественно, мужского пола) всегда получает от матери, поскольку только яйцеклетка передает свои митохондрии зарождающемуся эмбриону (митохондрии отцовской спермы не передаются).
Хотя подавляющее большинство пациентов с болезнью Лебера имеют гомоплазматические мутации, 10-15% мутаций – гетероплазматические.
За отличия в межиндивидуальных фенотипах может быть ответственна тканеспецифичная сегрегация. Некоторые исследования показывают, что риск для пациентов минимален, если гетероплазма составляет менее 60%.
Сыновья матерей, у которых уровни гетероплазмы ≤80%, с меньшей вероятностью будут страдать от болезни.
Обсуждаемый вопрос – появление синдрома Лебера у женщин-носителей мутаций, которые, в зависимости от генетического фона, имеют значительно меньшую пенетрантность, чем мужчины.
Некоторые исследования предполагают, что причина дифференцированной пенетрантности – модифицирующий Х-связанный ген, приводящий к проявлению заболевания у женщин только в гомозиготном состоянии.
Второй предполагаемый фактор – Х-инактивация «wild-type» Х-хромосомы.
Клиническая картина
Проявления нейропатии Лебера:
- внезапное безболезненное поражение обоих глаз;
- снижение остроты зрения;
- скотомы (темные пятна) в поле зрения;
- потеря цветового зрения;
- слепота;
- у женщин иногда наблюдаются симптомы, похожие на рассеянный склероз.
Шел 1994 год. В офтальмологическую клинику обратился 40-летний пациент с проблемой внезапной потери зрения на обоих глазах.
При составлении анамнеза врачи определили, что изначально произошла потеря зрения на одном глазу, затем – на втором. Постепенная слепота не сопровождалась болью.
Врачам пациент сообщил, что его брат (на 2 года младше) также несколько лет назад ослеп на один глаз.
Пациент прошел ряд обследований. Но все выводы были негативными, кроме выявления нарушения сердечного ритма. Также было исключено большинство офтальмологических диагнозов, которые могли бы объяснить безболезненную и быструю потерю зрения.
Так можно характеризовать клинический случай синдрома Лебера.
Диагностика и исследования
Подозрение на болезнь часто определяется окулистом или неврологом на основании анамнеза, оценки подробного осмотра глаз, состоящего из контроля остроты зрения, поля зрения, контрастной, цветовой чувствительности.
Золотой стандарт лабораторной диагностики – молекулярно-генетический анализ распространенных мутаций, проводимый из образцов крови или мазка из слизистых оболочек щек.
Это обследование проводится у пациентов с уже развитыми нарушениями зрения в рамках дифференциальной диагностики синдрома Лебера, или у бессимптомных членов семьи, еще не подвергавшихся сложному диагностическому процессу.
Но у бессимптомных пациентов молекулярно-генетическое тестирование не может предсказать развитие заболевания.
Для исключения распространенных мутаций целесообразно рассмотреть секвенирование генов мтДНК, кодирующих субъединицы в митохондриях, выделенных из мышечной биопсии.
Современные методы лечения
Лечение болезни Лебера – сложный процесс. Пациент должен отказаться от курения, максимально сократить употребление алкоголя, чтобы не повредить зрительный нерв. В терапии также используются некоторые витаминные и оксидазоснижающие соединения, но их эффект спорный.
До недавнего времени единственным вариантом облегчить течение болезни Лебера был коэнзим Q10, который посредством сукцинатдегидрогеназы обходит нефункциональный митохондриальный комплекс, увеличивая продукцию АТФ путем окислительного фосфорилирования.
Но это вещество обладает высокой липофильностью, и при пероральном введении его проникновение в митохондрии вызывает сомнения. Эффективность коэнзима Q10 никогда не была продемонстрирована в клинических исследованиях.
В последние годы было проведено несколько исследований для тестирования новых лекарственных средств. Предположительно, они положительно влияют на стабилизацию и восстановление зрительных функций. Особенно перспективны аналоги Убихинона с короткой цепью: Идебенон и α-токотриенолхинон (EPI-743), замещающие функцию дисфункционального комплекса.
Последствия и прогноз
Генетическая мутация приводит к нарушению функции зрительного нерва, вызывает нарушения зрения. Эти расстройства проявляются относительно рано – у старших подростков и молодых людей. Поражаются оба глаза, снижается острота зрения, в поле зрения могут появляться глазные капли с темными пятнами, переходящие в постоянное явление. Многие пациенты практически теряют зрение.
Профилактика
Поскольку атрофия зрительного нерва Лебера – это наследственная болезнь, ее профилактика сложная. В превентивных целях целесообразно вовремя лечить проблемы, способные стать причиной расстройства.
Следующий пункт – избегание любых ЧМТ, глазных травм. Важен также здоровый образ жизни, отказ от курения, потребления алкоголя.
Атрофия зрительного нерва Лебера
Наследственная оптическая нейропатия Лебера (Leber hereditary optic neuropathy, LHON), или наследственная атрофия зрительных нервов Лебера, или болезнь Лебера (не путать с амаврозом Лебера!!! – названия похожи, но клинические проявления различаются) –митохондриальное заболевание, манифестирующее, как правило, в возрасте 15-35 лет (однако возраст начала заболевания может варьировать от 1 до 70 лет). Леберовская атрофия зрительного нерва характеризуется острым или подострым двусторонним медленным снижением остроты центрального зрения, при этом не сопровождается болью в глазных яблоках. Глаза могут поражаться как одновременно, так и последовательно, с интервалом в несколько месяцев. Как правило, снижение зрения остается выраженным и постоянным, но описаны случаи, когда спустя несколько лет происходит спонтанное улучшение зрения, иногда значительное. На ранних стадиях заболевания часто отмечается поражение цветового зрения. В ряде семей кроме снижения остроты зрения выявляются и неврологические симптомы: тремор, атаксии, дистония, судороги, а в некоторых случаях – заболевания, не отличимые от рассеянного склероза. Характерными особенностями наследственной оптической нейропатии Лебера являются неполная пенетрантность (до 50% у мужчин и 10% у женщин) и большая частота заболевания среди мужчин (мужчины болеют в 3-5 раз чаще женщин), возможно, связанная с действием Х-сцепленного модифицирующего гена, расположенного в районе Xp21. Показано, что важное влияние на начало и развитие заболевания оказывают факторы риска – стрессы, курение, употребление алкоголя, действие токсинов, лекарств и инфекций.
Как и для других заболеваний с митохондриальным наследованием, для наследственной оптической нейропатии Лебера характерны передача по материнской линии, а также явление гетероплазмии (присутствие в клетке более одного типа митохондрий), которым в ряде случаев можно объяснить неполную пенетрантность.
Причиной развития наследственной оптической нейропатии Лебера являются мутации в митохондриальной ДНК. Выделяют 18 аллельных вариантов заболевания, связанных с миссенс-мутациями в ряде митохондриальных генов.
Большинство этих мутаций являются редкими (встречаются в одной или нескольких семьях в мире), однако в 95% случаев выявляется одна из трех мажорных мутаций: m.3460G>A, m.11778G>A или m.14484T>C.
Все они изменяют структуру генов, кодирующих белки первого комплекса дыхательной цепи митохондрий.
Показано, что степень тяжести заболевания и возможность восстановления зрения коррелируют с выявленными мутациями. Так, считается, что мутация m.11778G>A вызывает наиболее тяжелые формы, m.3460G>A – более легкие, а m.14484T>C дает наиболее благоприятный прогноз.
Нами разработан набор для ДНК-диагностики Атрофии зрительного нерва Лебера. Наборы предназначены для использования в диагностических лабораториях молекулярно-генетического профиля.
В Центре Молекулярной Генетики проводится диагностика основных трех мажорных мутаций m.11778G>A, m.14484T>C, m.3460G>A, а также 9 более редких первичных мутаций: m.3733G>A, m.4171C>A, m.10663T>C, m.14459G>A, m.14482C>G, m.14482C>A, m.14495A>G, m.14502T>C, m.14568C>T.