Фибродисплазия или болезнь Мюнхеймера (от лат. fibro – волокно, dis – расстройство, нарушение, plasis – строение, структура и os, ossis – кость, facio – делать; окостенение) – это патологическое состояние организма, среди всего населения планеты встречается очень редко, характерной чертой данного заболевания является перерождение мягкой скелетной ткани в костную.
Участки окостеневания формируются в месте повреждения мышечной ткани во время удара или ушиба, гематомы. Относится к группе редких болезней, частота заболеваемости 1:2000000.
На данный момент известно всего лишь около 600 человек, которые страдают данной патологией.
В связи с тем, что данная болезнь встречается довольно редко, ученым не предоставляется хорошая возможность изучить заболевания достаточно подробно.
Историческая справка
Впервые про оссифицирующую прогрессирующую фибродисплазию было упомянуто в 1648 году, когда один из докторов описал этот феномен, так как у него было так называемая «окаменевшая» пациентка. Более детально болезнь была описана уже в 1869 году немецким врачом, он не обнаружил связь между половыми признаками и расовой принадлежностью пациента, а также этнической предрасположенностью.
Патология начинает развиваться в детском возрасте, прогрессировать фибродисплазия продолжает в возрасте от 3-х до 4-х лет. Также ученым известны случаи, когда болезнь начала себя проявлять в трехмесячном возрасте.
Клинические проявления фибродисплазии
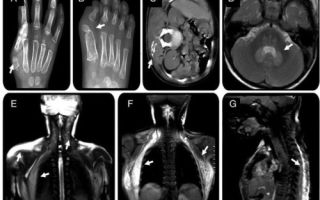
На данный момент существует только два клинических проявления, после которых можно поставить диагноз оссифицирующая прогрессирующая фибродисплазия. К ним относят: аномалия развития большого пальца, которая наблюдается с рождения ребенка и прогрессирующие гетеротопические окостенения на разнообразных участках тела.
Костные участки формируются поэтапно и впоследствии наносят необратимый вред здоровью человека. Пациенты с данной патологией в возрасте 30-ти лет уже вынуждены передвигаться на инвалидной коляске, и выполнять повседневные дела при помощи своих родных и близких.
Быстрая потеря в весе может привести к обездвиженности в области суставов, а также в результате образования костной ткани на стенках грудной клетки приводит к развитию сердечной недостаточности. Такие пациенты живут примерно до 40-ка лет.
Умирают они в основном от синдрома грудной недостаточности.
Причины развития заболевания
- травмирование кожных покровов;
- оперативное вмешательство;
- введение вакцины в организм человека;
- воздействие вирусных инфекций.
Диагностика фибродисплазии
Очень часто специалисты не могут поставить правильный диагноз. Поскольку не все специалисты обращают внимание на активно формирующиеся отеки мягких тканей в области шеи и головы, а также верхней части спины, наличие аномально развитого большого пальца ноги.
Очень редко только по одним клиническим проявлением можно диагностировать болезнь, обычно для этого нужно пройти рентгенографию, на которой будут обнаружены участки окостенения. Фибродисплазию чаще всего путают с саркомой мягких тканей, лимфедемой или агрессивным фиброматозом.
При проведении диагностики у маленьких детей часто проводятся ненужные анализы, а именно биопсия, которая дополнительно травмирует мягкие ткани и приводит к образованию окостеневших участков.
Стадии окостенения на рентгеновских снимках
При помощи рентгенографии можно определить стадию окостенения у пациента, они делятся на:
- Первая стадия (инфильтрация) характеризуется разрастанием молодой дегенеративной ткани, и образования вторичных дегенеративных изменений в мышечных структурах. На рентгеновских снимках эти изменения малозаметны.
- Вторая стадия или фиброзная индурация. Происходит рубцевание молодой ткани и развивается вторичная атрофия мышечных структур. На рентгеновских снимках можно увидеть нежные тени, напоминающие собой костную мозоль.
- Третья стадия или собственно окостенение. Это формирование собственно костной ткани на участках повреждения мягких тканей. На рентгеновских снимках видно очень четко участки с окостенением, а именно определяются крупные участки окостенения в мышечных тканях, которые срощены с костями, позвоночный столб похож на бамбуковую трость, и определяются анкилозы суставов.
Дополнительные аномалии развития скелета
Помимо больших пальцев аномальному развитию подвергается шейный отдел позвоночника. Жесткость в области шеи являются ранним диагностическим признаком фибродисплазии у пациентов. Дополнительно специалисты отмечают такие патологии: изогнутые, и в тоже время короткие пальцы, а также широкие кости бедер. Аномалии развития бедренных костей определяются на генетическом уровне.
Факторы развития фибродисплазии
Оссифицирующая фибродисплазия очень редкое заболевание, встречается с частотой 1:2000000. Было установлено что географические, гендерические, расовые и этнические факторы не влияют на развитие заболевания.
Чаще всего оссифицирующая фибродисплазия развивается из-за самопроизвольной мутации в генах. На дальнейшее развитие заболевания влияют экологические и генетические факторы.
При проведении исследования на близнецах было обнаружено, что под воздействием разных факторов окружающей среды фибродисплазия развивалась по-разному.
Стадии развития оссифицирующей прогрессирующей фибродисплазии
Выделяют несколько стадий развития процесса окостенения:
- Развитие небольшого участка воспалительного процесса, который ограничен в размерах.
- Прогрессирование болезни и задействование в воспалительный процесс близлежащих тканей.
- Образование бессосудистого уплотнения.
- Формирование хряща.
- Собственно окостенение.
Поэтапное развитие болезни
В зависимости от стадии, возраста и воздействия внешних факторов болезнь может вести себя по-разному. После завершения стадии обострения на протяжении определённого промежутка времени, если пациент не подвергался травмированию, фибродисплазия может не проявлять себя. Но обычно она активно прогрессирует, и в результате человек быстро теряет свою подвижность и становится прикованным к инвалидному креслу. Сначала формируются контрактуры в области локтевых и коленных суставов, что в свою очередь начинает ограничивать человека в движении. Далее подвергаются поражению более мелкие суставы и хрящи, которые соединяют ребра с позвоночником. Период, когда начинается стадия обострение заболевания невозможно предугадать, поскольку процесс дисплазии обычно запускает иммунная система. Практически все мышечные структуры организма подвержены окостенению.
Лечение оссифицирующей прогрессирующей фибродисплазии
Современная медицина еще не изобрела лекарство, которое лечило бы фибродисплазию. Но и методы по облегчению состояния больного значительных результатов не приносят.
При первых проявлениях заболевания положительная динамика наблюдается во время использования глюкокортикостероидов, тем самым уменьшая проявления болезни и улучшая качество жизни пациента, но к полному выздоровлению не приводит.
Полностью избавиться от костных наростов при помощи глюкокортикостероидов не представляется возможным.
Период обострения болезни начинается очень резко, даже спонтанно, и также неожиданно заканчивается, оставляя за собой костное образование. В современном мире тратится очень большое количество времени и средств на изучение данной патологии, целью данных исследований является улучшение качество жизни и состояния здоровья таких пациентов.
Было проведено исследование на лабораторных мышах, при использовании лекарственного препарата было установлено, что он помогает бороться с признаками фибродисплазия вызванной как мутировавшим геном, так и травмами.
На людях, к сожалению, эти испытания не проводились, потому что невозможно найти достаточное количество пациентов с фибродисплазией для проведения клинических испытаний. В виду отсутствия пациентов, данный лекарственный препарат будет находиться на стадии разработки еще длительное время.
Но в 2014 вроде удалось собрать необходимое количество пациентов для клинических испытаний лекарственного препарата «Паловаротен», испытания проводятся сразу в нескольких городах – Париж, Филадельфия и Сан-Франциско.
Среди клиник по лечению оссифицирующей прогрессирующей фибродисплазии самой лучшей считается лаборатория Маккея при Пенсильванском Федеральном Университете, которой руководит доктор медицинских наук Фредерик Каплан.
Это единственная в мире лаборатория, которая специализируется на изучение феномена фибродисплазии.
В России также можно провести диагностические процедуры относительно фибродисплазии, которые проводятся в медико-генетическом научном центре РАМН в Москве по ул. Москворечье д. 1.
В НИИ Ревматологии профессор Никишина И.П. и в ММА имени Сеченова профессор Голованова Н.Ю. (Москва) проводят терапевтическое лечение, а также в Иркутском государственном медицинском университете доктор медицинских наук Калягин А.Н.
(Иркутск) и в Казанской Государственной Медицинской Академии доктор медицинских наук Мальцев С.В. (Казань).
Профилактические мероприятия для улучшения качества жизни у больных с фибродисплазией
При соблюдении некоторых простых правил можно избежать быстрого прогрессирования болезни. Для этого необходимо избегать больных с ОРВИ, травмирования тканей, перегревания, а также переохлаждения организма.
Это не гарантирует полную защиту от прогрессирования болезни, но приостановит ее на некоторое время.
Своевременная диагностика и правильная постановка диагноза позволяет начать раннюю терапии глюкокортикостероидами, и тем самым продлить жизнь пациенту и улучшить качество его жизни.
Также в профилактических целях можно использовать Интерферон человеческий лейкоцитарный. Во время длительной терапии интерфероном понижается количество мышечных и подкожных инфильтратов. Также возможно использование биофосфонатов и этилендиаминтетрауксусной кислоты.
Прогноз
К сожалению, исход данного заболевания является не очень благоприятным для пациента. Продолжительность жизни таких пациентов очень разнообразна. Все зависит от воздействия внешних факторов и генетических модификаций.
Источник: https://health-post.ru/fibrodisplaziya/
Фибродисплазия, что такое и как лечится фибродисплазия оссифицирующая прогрессирующая?
Фибродисплазия является патологическим процессом, который встречается достаточно редко и характеризуется непрерывным окостеневанием мягкой ткани скелета. Подобная болезнь способна затрагивать соединительные ткани в мышцах, апоневрозы, фасции и сухожилия.
Наблюдается преимущественно в детском возрасте в качестве болевых, похожих на опухоль, образований, локализующихся в мягких тканях. Фибродисплазию ассоциируют с врожденными отклонениями в развитии скелета.
Заболеванию свойственны эпизодические вспышки напухания в мягких тканях, что провоцирует их преобразование в костную ткань.
Причины
Фибродисплазия оссифицирующая прогрессирующая является болезнью, первопричиной которой станут нарушения в образовании костной ткани. Патология зарождается на этапе развития внутри утробы, когда мутирует ген ACVR1, расположенный во 2 хромосоме.
Вследствие подобной реакции формируется ненадлежащая форма белка, отвечающая за рост костей. Он возникает в остеобластах, а также в волокнах рубцовой ткани. Последующее развитие патологического процесса фактически не исследовано.
Очаги рассматриваемой болезни зачастую локализуются там, где были повреждены какие-либо мягкие ткани – ушиб, порез, укус, ожог. В такой ситуации присутствует значительное содержание белых кровяных телец, что станет причиной воспалительных или иммунных изменений.
Мутация гена ACVR1 провоцирует возникновение данной патологии у ребенка. Она также несет ответственность за определенные врожденные пороки формирования скелета. Зачастую подобными проявлениями станут: микродактилия, варусная кривизна большого пальца ступни, сращивание шейных позвонков, сращивание реберно-позвоночного сустава.
Вследствие того, что костные поражения тесным образом взаимосвязаны с мышечной слабостью, у больных с такой болезнью часто отмечается кривизна позвоночного столба, что провоцирует различные неврологические сложности.
Подобный патологический процесс считается врожденным наследственным заболеванием. Для него своейственно стремительно прогрессирующее протекание, вследствие чего у пациента отмечаются расстройства в опорно-двигательной системе. Люди становятся глубокими инвалидами и умирают преимущественно в детском либо молодом возрасте.

Симптомы
Симптоматика заболевания, которое также носит название болезни Мюнхеймера нечасто проявляется сразу после появления на свет. Специалисты делают акцент лишь на нарушенном формировании пальцев на верхних и нижних конечностях малыша.
Какие-либо другие проявления во время внешнего осмотра отсутствуют. Начинается патология внезапно без каких-то объективных провоцирующих факторов. Возраст ребенка варьируется от 2 месяцев до 3-5 лет.
Ранняя симптоматика – возникновение уплотнений в шейных, спинных и мышцах рук. Их габариты бывают от 3 мм до 10 см.
При пальпации отмечается незначительный дискомфорт, кожный покров над этой областью зачастую протекает без патологических изменений, изредка возникает краснота. Подобные образования расположены в местах ушибов, травматизма, порезов и прочих поражений.
Походка во время наследственной фибродисплазии станет неустойчивой и скованной. Лицо фактически не будет выражать какие-либо эмоции, шейные и спинные мышцы находятся все время в напряжении. Со временем снижается двигательная активность в суставах, что сопряжено с возникновением новых уплотнений. На терминальном этапе формирования больной станет фактически в полной мере обездвиженным.
При сдавлении нервных окончаний спинного мозга отмечаются жалобы на болезненные ощущения в области спины, верхних и нижних конечностях, а также на сбой в восприимчивости кожного покрова. Рассматриваемый патологический процесс обладает симптоматикой, выраженность которой увеличивается ежедневно.
Известны случаи окончательного рассасывания новообразований, однако далее они возникали вновь. По прошествии 15-20 суток с момента образования узла он станет кальцинатом, будет напоминать костную ткань и рассосаться не сможет.
У взрослого бывает до 20-30 подобных очагов. Они сливаются в одно целое и образуют «второй скелет». Подобное ведет к достаточно скорой инвалидности.
Со стороны внутренних органов наблюдаются разнообразные расстройства, что вызывает смертельный исход.
Кроме того, симптомы отображают отклонения в формировании человека либо окостенения тканей, к примеру:
- короткий 1 палец ступни;
- клинодактилия (отсутствует сустав 1 пальца ступни, изогнут внутрь);
- недоразвитость фаланги;
- искривленный 5 палец;
- отечность в мягких тканях головы от наименьшего поражения (незначительный ушиб, ссадина);
- отклонения в структуре шейных позвонков;
- более широкие и короткие кости в плечах и бедре;
- вероятна задержка психического развития, глухота.
Зачастую фибродисплазия путается с онкологическими заболеваниями и прочими патологиями, во время которых образуются различные затвердевания. Специалисты пытаются устранить их, но подобное ведет только к ускорению формирования дополнительных костных новообразований.
Виды
Четкие перемены внутри человеческого организма обнаруживаются на результатах рентгенологической диагностики. Кроме того, выделяются следующие разновидности окостенения:
- параартикулярная;
- параоссальная;
- травматическая.
Кости, появившиеся подобным образом, не отличаются от прочих костей внутри человеческого организма.
Оссифицирующая фибродисплазия может быть классифицирована по этапам окостенения:
- первый – увеличивается количество молодой дегенеративной ткани, возникают вторичные деструктивные процессы в мышцах;
- второй – развивается атрофия мышц, рубцуются молодые ткани;
- третий – формируется костная ткань на тех участках, где отмечалось повреждение мягкой ткани.
Исходя из этого, проведя анализ этапов патологического процесса, возможно утверждать, что различные области, поврежденные рассматриваемым заболеванием с разной скоростью окостеневают. Наряду с этим, невзирая на то, что подобное костетворение похоже на костеобразование скелета у зародыша, основным отличием станет то, что в первом случае отсутствует воспалительный процесс.
Однако, специалисты едины во мнении, что такие различия являются условными, поскольку точно не подтверждаются вследствие того, что биопсия (забор фрагмента поврежденных участков тела) у таких пациентов может спровоцировать расширение воспалительного очага.
Диагностика
Патологический процесс допустимо обнаружить лишь после форсированного образования костей в тканях скелета, которое проявляется у детей, с момента появления на свет и до 13-14-летнего возраста. Чаще всего заболевание отмечается на 2,5 году после появления болезни. Однако, отклонения врожденного характера, обращают внимание сразу после родов.
Диагностические меры осуществляются следующими способами:
- Наружный осмотр.
- Рентгенография.
- Генетическое исследование.
Описание наружного осмотра варьируется от возрастных показателей малыша. Таким образом, к примеру, до 10 – 12 лет главными признаками болезни станут множественные уплотнения, располагающиеся фактически на каждом участке тела.
У детей более старшего возраста, помимо указанной симптоматики, возникает ограниченная двигательная активность суставов, кривизна позвоночного столба и многочисленные очаги окостенения.
Подобные новообразования возможно обнаружить невооруженным глазом, что существенно обезображивает тело.
В определенных ситуациях проводят гистологическую диагностику подобных очагов, однако осуществляется подобное нечасто, поскольку поражение кожного покрова провоцирует появление возникновение новых оссификатов.
Рентгенография может быть полезной на предварительно стадии формирования заболевания, поскольку посредством нее возможно обнаружить такие патологические процессы, как клинодактилия либо костная дисплазия.
Дальше в сухожилиях, фасциях и в мышцах образуются уплотнения, носящие костное строение.
Когда болезнь находится на запущенной стадии, то данные уплотнения не дадут возможности увидеть на снимках внутренние органы.
Генетическая диагностика осуществляется в целях того, чтобы обнаружить мутацию в гене ACVR1. Такой анализ не нуждается в особой подготовке: достаточным будет сдать кровь на голодный желудок и на протяжении 7 суток получить результаты.
Если присутствует рассматриваемая патология специалисты во всех случаях используют рентгенографическую методику, так как с ее помощью выявляются костные уплотнения.
Когда же фибродисплазия станет выраженной, то за новообразованиями возможно не увидеть внутренние органы.
На сегодняшний день точно не установлены гены данной болезни. Допустимо сделать предположение, что фибродисплазия, вывяленная у больного, закладывается генетически. Кроме того, существует риск того, что заболевание бывает спровоцировано несколькими генами, воздействующими едино.
Лечение
На данный момент медицина не дает предложений о действенных терапевтических методиках подобной патологии. Лечение, проводимое для устранения оссификатов, проводится крайне часто. Однако после удаления старых наростов образуются новые, большие по размерам. Потому оперативное вмешательство не может быть действенным. Консервативное лечение проводят непосредственно симптоматическое.
Одним из эффективных средств, которые обнаружены врачами, считается Этидронат. Когда пациент получит большую дозировку лекарства, болезнь замедлится в росте.
Однако в большом количестве медикамент может разрушать, кроме костных новообразований, основной скелет. Некоторые специалисты рекомендуют использовать средства, которые содержат памидроновую кислоту.
Препарат воздействует аналогичным образом с этидронатом, но пагубного воздействия на скелет оказывает значительно меньше.
Остальная терапия направлена на улучшение самочувствия больного при обострении фибродисплазии, а в этих целях применяют такие лекарства:
- Преднизолон. Дает антивоспалительный эффект, который призван устранить болезненные ощущения. Употребляется в течение 15 дней.
- Целебрекс. Способствует замедлению воспалительных изменений внутри организма. Назначает врач по рецепту, поскольку полностью не изучены побочные эффекты от препарата.
- Монтелукаст. Используется совсем нечасто, поскольку воздействие лекарства окончательно не установлено.
- Метаквалон. Уменьшает неприятные ощущения во время обострения патологического процесса. Поскольку, не подтверждаются сведения о безвредности лекарства, курс подбирают в индивидуальном порядке для каждого больного.
- Употребление медикаментов помогает пациентам легче устранить очередную вспышку патологического процесса.
- Вследствие того, что фибродисплазия наблюдается очень редко, помимо этого, она характеризуется проявлениями различной тяжести, ожидаемые экспериментальные терапевтические методики проходят серьезную оценку относительно их дозы и продолжительности лечебного курса.
Осложнения и последствия
Прогрессирующая фибродисплазия способна протекать молниеносно, ведет к опасным расстройствам опорно-двигательной системы, инвалидности и преждевременному смертельному исходу. Наблюдается зачастую в детском возрасте в качестве болезненных, похожих на остит, наростов, локализующихся в мягких тканях.
Возникновение подобных новообразований бывает спровоцировано внутримышечными инъекциями, получениями закрытых травм, оперативным вмешательством. Но рассматриваемый патологический процесс способен сформироваться и без объективных причин. В основном после оперативного удаления наростов оссифицирующая фибродисплазия продолжит прогрессировать.
Процент выживаемости крайне низок.
Профилактика
Достаточно соблюдать определенных нетрудных предписаний, чтобы по максимуму купировать развитие рассматриваемой патологии:
- требуется предотвратить контактирование с инфицированными ОРЗ, ОРВИ;
- предупредить травматизм;
- исключить переохлаждание и перегрев.
Естественно, гарантировать окончательное восстановление в такой ситуации нельзя, однако замедлить развитие болезни подобным методом возможно. Кроме того, в терапевтических целях допустимо использовать лейкоцитарный Интерферон для уменьшения числа подкожных и мышечных инфильтратов.
Прогноз подобного патологического процесса считается неблагоприятным, поскольку длительность жизни пациентов крайне разнообразна. Многие аспекты варьируются от провоцирующих факторов.
Фибродисплазия считается прогрессирующей оссифицирующей редкой генетической болезнью, которая вызвана мутацией гена ACVR1. Патологии характерны отклонения врожденного характера в структуре скелета.
Они спровоцированы окостенением рубцовой ткани, мышц, сухожилий в определенных отделах. В результате прогрессирования заболевания у пациентов отмечаются сбои в опорно-двигательной системе.
Действенная терапия, которая способна окончательно избавить от заболевания, отсутствует.
(1
Источник: https://orto-cure.ru/chto-takoe-fibrodisplazija-i-kak-ee-lechit/
Оссифицирующая прогрессирующая фибродисплазия (ОПФ)
Что такое оссифицирующая прогрессирующая фибродисплазия?
Оссифицирующая прогрессирующая фибродисплазия (или сокр. ОПФ, болезнь Мюнхеймера, синдром каменного человека) — очень редкое наследственное заболевание, при котором соединительные ткани тела, включая мышцы, сухожилия и связки, постепенно замещаются костью (в процессе, называемом оссификацией).
Болезнь Мюнхеймера присутствует при рождении, однако симптомы могут не проявляться до раннего детства. Окостенение может произойти случайно или после травмы.
Симптомы ОПФ
У рожденных с ОПФ, признаки и симптомы окостенения могут никак не выявляться, пока ребенок не станет немного старше и не начнет расти.
У новорожденных первым признаком ОПФ часто является врожденная аномалия пальцев ног.
Вскоре после рождения медицинские работники или родители могут заметить, что большие пальцы ноги ребенка короче других пальцев (см. фото выше).
Этот порок развития наблюдается у всех людей с синдромом каменного человека и является важной подсказкой для постановки диагноза.
У новорожденного также могут быть отеки вокруг глаз и кожи головы.
В некоторых случаях этот отек может начаться, когда плод еще находится в утробе матери, хотя это состояние обычно диагностируется только после рождения.
Около 50 процентов людей с этим заболеванием также имеют сходные врожденные аномалии в больших пальцах — также наблюдались и другие пороки развития, например, в позвоночнике.
Большинство людей с оссифицирующей прогрессирующей фибродисплазией впервые испытывают основные симптомы заболевания (иногда их называют «обострениями») к 10 годам жизни.
В то время как общая скорость прогрессирования состояния неизвестна, окостенение имеет тенденцию следовать определенной схеме, начиная с шеи и вплоть до плеч, туловища, конечностей и ступней.
Однако, поскольку на формирование кости могут повлиять травма (например, перелом руки) или вирусное заболевание (например, грипп), заболевание может не строго следовать этому прогрессу.
Основные симптомы болезни Мюнхеймера зависят от того, какие части тела стали окостеневшими. Часто, при заболевании встречаются нежные комки под кожей (подкожные узелки, см. фото выше).
Иногда формированию этих узелков предшествует умеренная лихорадка.
Большинство людей с ОПФ будут иметь общие симптомы боли, ригидности и прогрессирующего недостатка подвижности по мере разрастания костной ткани.
В зависимости от того, какие части тела окостенеют, более специфические симптомы ОПФ могут включать:
- проблемы с питанием, которые могут привести к дефициту питательных веществ или недоеданию;
- трудно с разговором;
- проблемы с зубами;
- затрудненное дыхание;
- респираторные инфекции;
- нарушение слуха;
- выпадение волос (облысение);
- анемию;
- сдавливание нервов;
- правосторонняя застойная сердечная недостаточность;
- искривление позвоночника (сколиоз и кифоз).
- сенсорные аномалии;
- умеренную умственную отсталость;
- неврологические симптомы.
X-сцепленная миопатия с чрезмерной аутофагией
Люди с синдромом каменного человека могут иметь периоды в своей жизни, когда костная ткань не будет расти. В других случаях может показаться, что это явление происходит случайно и при отсутствии каких-либо очевидных травм или заболеваний. Если оссификация происходит в необычной части тела (где кости обычно не обнаруживаются), это может привести к переломам.
Со временем формирование новой опухоли костей и тканей, которая сопровождает это состояние, может серьезно повлиять на то, насколько хорошо человек может двигаться.
В большинстве случаев оссифицирующая прогрессирующая фибродисплазия в конечном итоге приводит к полной иммобилизации. Многие люди с этим заболеванием к 30 годам будут прикованными к постели.
Причины фибродисплазии
У большинства людей, рожденных с ОПФ, заболевание возникает в результате случайной генетической мутации. Хотя это генетическое заболевание, оно обычно не встречается у всей семьи.
Человеку нужен только один пораженный ген, чтобы у него возникла оссифицирующая прогрессирующая фибродисплазия. Большинство случаев происходит из-за случайной мутации — у человека редко развивается состояние, потому что только от одного из родителя наследуется ненормальный ген. В генетике это явление называется как аутосомно-доминантное расстройство.
Мутация гена, ответственная за состояние, была идентифицирована исследователями из Университета Пенсильвании — они идентифицировали рецептор на хромосоме 2, называемый рецептором активина типа IA (ACVR1). ACVR1 присутствует в гене, который кодирует костные морфогенетические белки (BMP), которые помогают формировать и восстанавливать скелет, начиная с момента образования эмбриона.
Исследователи полагают, что мутация в гене предотвращает отключение этих рецепторов, что позволяет неконтролируемой кости образовываться в тех частях тела, где она обычно не появляется в течение жизни человека.
Диагностика
Оссифицирующая прогрессирующая фибродисплазия встречается очень редко.
Предполагается, что только несколько тысяч человек страдают этим заболеванием, и в мире существует только около 800 известных пациентов с этим заболеванием — 285 из них находятся в Соединенных Штатах.
По-видимому, ОПФ чаще встречается у детей определенной расы, и такое состояние встречается у мальчиков так же часто, как и у девочек.
Диагностика оссифицирующей прогрессирующей фибродисплазии может быть затруднена. Нередко изначально это заболевание ошибочно диагностируется как форма рака или как состояние, называемое агрессивным ювенальным фиброматозом.
В начале диагностики болезни Мюнхеймера, если ткань подвергается биопсии и исследованию под микроскопом (гистологическое исследование), она может иметь некоторые сходства с агрессивным ювенильным фиброматозом. Однако при последнем заболевании поражения не прогрессируют до полностью сформированной кости, как при синдроме каменного человека. Это может помочь врачу провести различие между данными болезнями.
Фибромиалгия: что это такое, причины, симптомы и лечение
Одним из основных диагностических признаков, который может привести врача к подозрению на болезнь Мюнхеймера, в отличие от другого состояния, является наличие коротких, неправильно сформированных больших пальцев ног.
Если биопсия ткани неясна, клиническое обследование ребенка может помочь врачу исключить агрессивный ювенильный фиброматоз. У детей с агрессивным ювенильным фиброматозом нет врожденных пороков развития пальцев ног или рук, однако она почти всегда встречается у детей с ОПФ.
Другое состояние, прогрессирующая костная гетероплазия, может также быть перепутана с оссифицирующей прогрессирующей фибродисплазией. Основное различие при постановке диагноза заключается в том, что рост кости при прогрессирующей костной гетероплазии обычно начинается на коже, а не под ней. Эти костные бляшки на поверхности кожи отличаются от нежных узелков, которые образуются при ОПФ.
Другие методы обследования, которые доктор может назначить, если есть подозрения на синдром каменного человека, включают:
- полный медицинский анамнез и медицинский осмотр;
- рентгенологические исследования, такие как компьютерная томография (КТ) или сцинтиграфия костей (сканирование костей) для поиска изменений скелета;
- лабораторные анализы для измерения уровня щелочной фосфатазы;
- генетическое тестирование для поиска мутаций.
Если подозревается наличие ОПФ, врачи стараются избегать любых инвазивных тестов, процедур или биопсии, потому что травма обычно приводит к образованию большего количества костной ткани у человека с таким заболеванием.
Хотя состояние обычно не встречается у всей семьи, родителям, у которых есть ребенок с диагнозом болезни Мюнхеймера, может быть полезна медико-генетическая консультация.
Лечение
В настоящее время нет лекарства от ОПФ. Также нет определенного или стандартного курса лечения. Существующие методы терапии не эффективны для каждого пациента, и поэтому основной целью является лечение симптомов и предотвращение роста костей, если это возможно.
Хотя терапия не остановит прогрессирование состояния, медицинские решения по лечению боли и других симптомов, связанных с ОПФ, будут зависеть от потребностей конкретного пациента. Врач может порекомендовать попробовать один или несколько из следующих способов лечения препаратами и процедурами, чтобы улучшить качество жизни пациента:
- Преднизон в высоких дозах или другой Кортикостероид;
- такие препараты, как Ритуксимаб (обычно используется для лечения ревматоидного артрита);
- Ионтофорез лекарственных веществ для доставки лекарств через кожу;
- Миорелаксанты или инъекции местного анестетика;
- препараты под названием Бисфосфонаты, которые используются для защиты плотности костей;
- Нестероидные противовоспалительные препараты (НПВП);
- лекарства для подавления тучных клеток, которые могут помочь уменьшить воспаление.
Окостенение часто происходит случайным образом и не может быть полностью предотвращено, однако, в редких случаях, оно также может происходить в ответ на воспаление, травму и болезнь.
Таким образом, рекомендации относительно активности, образа жизни, профилактического ухода и вмешательств могут быть назначены начиная с детства.
Антифосфолипидный синдром
Рекомендации могут включать:
- избегать ситуаций, которые могут привести к травме, например, полное исключение занятий спортом;
- избегать инвазивных медицинских процедур, таких как биопсия, стоматологических процедур и внутримышечной иммунизации;
- профилактика антибиотиками для защиты от болезней или инфекций, когда это необходимо;
- меры по профилактики инфекций, например, надлежащая гигиена рук, для защиты от распространенных вирусных заболеваний (таких как грипп) и других респираторных вирусов, а также от осложнений, таких как пневмония;
- физиотерапия;
- средства передвижения и другие вспомогательные устройства, такие как ходунки или инвалидная коляска;
- другие медицинские устройства, которые могут помочь в повседневной жизни для переодевания и купания.
- медицинские устройства или другие меры безопасности для предотвращения падений, например, при вставании с постели или приеме душа;
- психологическая и социальная поддержка пациентов и их семей;
- образовательная поддержка, включая специальное образование и обучение на дому;
- для семей может быть полезным медико-генетическая консультация.
Инвазивные процедуры или операции, направленные на удаление областей аномального роста кости, не рекомендуются, так как травма от операции почти всегда приводит к развитию дальнейшей окостенения.
Если операция абсолютно необходима, следует использовать наиболее минимально инвазивную методику. Пациентам с ОПФ также могут потребоваться особые анестезирующие средства.
В последние годы было проведено несколько клинических испытаний с целью разработки лучших вариантов лечения для людей с фибродисплазией.
Резюмируя
Оссифицирующая прогрессирующая фибродисплазия является чрезвычайно редким состоянием, при котором мутация гена заставляет соединительные ткани тела, включая мышцы, сухожилия и связки, постепенно замещаться костью (процесс называемый окостенением).
От болезни Мюнхеймера не существует лекарств и диагностика состояния довольно сложна.
Лечение в основном поддерживающее, и прогрессирование состояния обычно довольно непредсказуемо. Принятие мер во избежание травм и других ситуаций, которые могут привести к усилению окостенения, может помочь уменьшить количество замещений у человека, однако новая кость может все еще образовываться без какой-либо явной причины.
ОПФ обычно приводит к полной неподвижности, и к 30 годам большинство людей приковано к постели. В настоящее время проводятся клинические испытания, которые, как мы надеемся, позволят найти лучшие варианты лечения для повышения качества жизни пациентов с этим заболеванием.
Видеозаписи по теме
Источник: https://tvojajbolit.ru/revmatologiya/ossifitsiruyushhaya-progressiruyushhaya-fibrodisplaziya-opf/
Фибродисплазия: причины, симптомы, диагностика и лечение
В этой статье мы расскажем вам о тяжелейшем инвалидизирующем заболевании, которое встречается очень редко — 1 человек на 2 млн населения. Всего в мире насчитывается около 500 больных этим недугом.
Он заключается в том, что мышцы больных постепенно превращаются в кость, а сам человек постепенно, по сути, становится статуей и утрачивает возможность двигаться. Название этого недуга — фибродисплазия.
Давайте узнаем, из-за чего развивается эта болезнь, кто ей подвержен и реально ли ее вылечить.
Что такое фибродисплазия?
У новорожденных можно с 95% вероятностью определить ПОФ — большой палец ноги загнут внутрь и зачастую там отсутствует сустав. Обычно болезнь проявляет себя в течение первых 10 лет жизни ребенка и совершенно внезапно. У детей все начинается с шеи и постепенно спускается до нижних конечностей.
Скорость развития также невозможно установить точно, но чем больше травм получает больной и чем больше хирургических вмешательств происходит, тем быстрее развивается недуг.
Довольно часто ПОФ путают с раком и прочими болезнями, в результате которых могут развиться подобные костные образования. Врачи удаляют эти образования, но, к сожалению, это приводит к усугублению ситуации и ускорению развития фибродисплазии.
В этой передаче рассказывается о двух больных девушках (русской и американки). Они рассказывают о том, как все началось и возможно ли вылечить ПОФ. У американской девушки врачи первоначально диагностировали рак и ампутировали больную руку в раннем детстве, и только потом выяснилось, чем она в действительности больна.
Причины
Что именно запускает мутацию на генном уровне, ученые пока не могут выяснить. На данный момент известно только то, что это наследственный недуг и передается аутосомно-доминантным путем.
Это значит, что у больного ПОВ 100% будут дети с точно такой же патологией. Однако люди с болезнью Мюнхеймера редко доживают до возраста, когда могут рожать детей.
А если и доживают, то женщинам смертельно опасно беременеть из-за изменений, происходящих в организме. Так что ПОФ является итогом индивидуальных генетических мутаций.
Только в 2006 году ученые выявили, мутация какого именно гена причастна к развитию фибродисплазии и сейчас проходят исследования, чтобы создать препарат, позволяющий лечить этот недуг.
Зачастую у носителей мутировавшего гена недуг никак не показывает себя до какого-то времени. Толчком к развитию могут послужить:
- травмы,
- вакцинации,
- вирусное заражение,
- хирургические вмешательства.
Симптоматика
Самыми очевидными симптомами болезни Мюнхеймера является окостенение мягких тканей, но кроме этого могут быть и другие дефекты, указывающие на возможное наличие недуга:
- короткий изогнутый внутрь большой палец стопы,
- дефекты в позвоночнике, особенно в шейном отделе,
- короткий большой палец на руках,
- возможно облысение, глухота, замедленное развитие,
- искривление или скос пятого пальца.
Достаточно часто ПОФ провоцирует развитие кривошеи, сколиоза и тугой подвижности костных сочленений.
Если вы заметили у ребенка подобные признаки недуга, то обратитесь к врачу. Доктор, если посчитает нужным, назначит генетическое исследование, в результате которого либо оправдаются подозрения на наличие генной мутации, либо нет.
Диагностика и лечение
Как мы уже упоминали выше, зачастую на ранней стадии фибродисплазии врачи ставят неверный диагноз, что в свою очередь только усугубляет развитие недуга. Обычно болезнь Мюнхеймера диагностируют в возрасте около 14 лет, когда все симптомы становятся явными.
Генетический анализ — единственный способ точно определить наличие изменений в ДНК.
К сожалению, пока болезнь Мюнхеймера считается неизлечимой. Однако американские ученые, проводя исследования, уже выявили, как мы говорили, ген ACVR1, при мутации которого происходит развитие недуга. Сейчас продолжается работа над генными блокаторами, которые должны остановить мутацию. Из-за того, что это заболевание очень редкое довольно сложно проводить исследования.
Несмотря на это, в 2011 году была разработана особая РНК-молекула, которая может убрать поврежденную копию гена ACVR1, никак не влияя на его нормальную копию.
Чтобы проверить, насколько эффективен данный способ, были проведены проверки на стволовых клетках, которые были получены из молочных зубов больных фибродисплазией.
В клетках были измененные и нормальные копии ACVR1/ALK2 рецептор-белков.
В ходе опыта ученые выяснили, что созданная мРНК способна восстанавливать нормальный синтез белков такой же, как у здоровых людей, нейтрализовав мутировавший ген.
В этом видео рассказывается о том, каким образом проводятся эти анализы и как в принципе выявляются генетические заболевания.
Еще 10 лет назад было невозможно представить, что этот недуг излечим. Сейчас у страдающих болезнью второго скелета появилась надежда на здоровую и счастливую жизнь.
Конечно, предстоит еще большая работа, этот подход к лечению нужно опробовать на лабораторных мышах и только потом перейти к испытаниям на людях. Но, тем не менее, это уже большой шаг. Будем надеяться, что исследования закончатся успешно.
Источник: https://prosustav.ru/arthropathy/displaziya/fibrodisplaziya-bolezn-myunxejmera.html
Причины развития, симптомы и лечение фибродисплазии
Фибродисплазия, или болезнь Мюнхеймера, а также «болезнь второго скелета» — невероятно редкое и очень тяжёлое по своему течению заболевание генетической природы. В основе его — постепенное превращение мышц, сухожилий и связок в кости. Название патологии можно перевести как «мягкая соединительная ткань, которая постепенно в течением времени превращается в кость».
Немного истории
С годами процесс постепенно прогрессирует, а сама патология даёт о себе знать в первые десять лет жизни ребёнка, при этом выявляется мутация определённого гена. Фибродисплазия была известна очень давно и регистрировалась в XVII и XVIII веках.
Первое упоминание можно встретить в письме врача-француза, в 1692 году, однако первое подробное описание патологии было произведено в 1736 году. Сделал это врач Джон Фрек, который на протяжении довольно длительного времени наблюдал одного подростка. Болезнь получила название «миозит оссифицирующий прогрессирующий».
Современное название патология получила в 1970 годах. Изменил его ведущий сотрудник школы медицины и медицинской генетики. Тогда же стало понятно, что это врождённая наследственная патология, которая характеризуется прогрессирующим течением, нарушением функций опорно-двигательного аппарата, инвалидностью и смертью, преимущественно в детском или подростковом возрасте.
Причина
В основе заболевания лежит воспалительный процесс сухожилий, связок, фасций и других составляющих опорно-двигательного аппарата. В итоге наступает их кальцификация и окостенение. Вместо обычных противовоспалительных реакций организм начинает реагировать необычным явлением – начинают расти кости там, где их быть не должно.
Единственный в мире институт, который занимается только этим заболеванием – это лаборатория Маккея.
Клиническая картина
Прогрессирующая оссифицирующая фибродисплазия проявляется с первых дней после рождения. Основной признак – это патология большого пальца ноги. При осмотре видно, что одна или несколько фаланг пальчика искривлены, а иногда здесь же не хватает сустава. Именно такой палец на 95% всех случаев и даёт возможность подозревать описываемую патологию.
Ещё одна особенность этого генетического заболевания – обострения. Самая распространённая картина этого периода – это появление под кожей уплотнений, которые можно прощупать руками. Они могут появиться практически на любом участке тела. На месте таких уплотнений постепенно появляются оссификаты, которые не поддаются никакому лечению.
Ещё один симптом – отёк мягких тканей головы, что часто бывает при травмах. Он может держаться на протяжении нескольких месяцев, не спадает и не поддаётся никакому лечению.
Некоторые путают это заболевание с раковыми опухолями и даже пытаются удалить эти шишки, однако это приводит к ещё более бурному росту «лишних» костей. Именно это приводит к инвалидизации пациента.
После исследования оссифицирующей фибродисплазии стало понятно, что некоторые ткани никогда не затрагиваются патологическими оссификатами. К таким мышцам можно отнести гладкую и сердечную мускулатуру. При этом сам процесс формирования таких костей не причиняет каких-то неприятных ощущений, хотя некоторые могут жаловаться на незначительные болевые ощущения и повышение температуры тела.
Диагностика
Основной способ диагностики – рентгенографический. Оссификаты могут быть как единичными, так и сливаться в обширные очаги. Основное расположение – боковая поверхность грудной клетки и плечи.
При этом во время исследования анализа крови и анализа мочи нет каких-либо отклонений. То же самое касается анализа на гормоны, и показателей минерального обмена.
Однако необходим обязательный генетический анализ, который позволяет поставить единственно правильный диагноз, так как деформация первых пальцев стоп часто диагностируется как вальгусная деформация и ей не уделяется достаточного внимания в лечении.
Что же касается профилактики заболевания у плода, то в связи с отсутствием данных о том, что может стать причиной развития этого заболевания, предупреждение заболевания сильно затруднено.
Консервативное лечение
На сегодняшний день известно не более 800 случаев прогрессирующей фибродисплазии, однако до сих пор не найдено ни действенной профилактики, ни эффективного метода лечения. Такая терапия, как удаление оссификатов, проводится довольно часто, однако на их месте в самый короткий срок появляются новые, ещё большие по размеру. Поэтому назвать оперативное лечение действенным здесь нельзя.
Что же касается консервативной терапии, то она проводится симптоматически.
Источник: https://nashynogi.ru/raznoe/prichiny-razvitiya-simptomy-i-lechenie-fibrodisplazii.html
Прогрессирующая оссифицирующая фибродисплазия
Прогрессирующая оссифицирующая фибродисплазия (ПОФ) (от лат.
fibro – волокно, dis – расстройство, нарушение, plasis – строение, структура и os, ossis – кость, facio – делать, окостенение) – редкое аутосомно-доминантное заболевание, характеризующееся прогрессирующим гетеротопическим истинным окостенением мягких тканей скелета (соединительнотканных прослоек в толще мышц, фасций, апоневрозов, сухожилий) и ассоциирующееся с множественными врожденными скелетными аномалиями. В МКБ-10 ПОФ представлена отдельной рубрикой – M 61.1.
ПОФ – это редкая нозологическая форма, характеризующаяся рецидивирующими болезненными эпизодами припухлости мягких тканей, ведущими к гетеротопической оссификации через стадию эндохондрального процесса, т. е. превращением соединительной ткани межмышечных прослоек, сухожилий и связочного аппарата в костную ткань [1, 2].
В литературе это заболевание можно встретить под названиями: параоссальная гетеротопическая оссификация, болезнь Мюнхмайера, «болезнь второго скелета», опухоль Стернера [3, 4].
Наиболее часто употреблявшееся ранее название – «оссифицирующий миозит» («превращение мышцы в кость») в настоящее время считается некорректным, поскольку не вполне отражает патогенетические и патоморфологические основы болезни.
Общепризнанным научным термином, обозначающий данную патологию, является термин «прогрессирующая оссифицирующая фибродисплазия», что указывает на вовлечение в процесс окостенения соединительнотканных структур вследствие особого характера воспалительных процессов в сухожилиях, связках, фасциях, апоневрозах и мышцах, заканчивающихся окостенением. Расовой, половой и географической предрасположенности к ПОФ не обнаружено [5].
Интерес к этому заболеванию существует с давних времен. Первое упоминание о ПОФ относится к 1648 г., когда Patin описал «окостеневшую» пациентку [6]. В 1996 г. R. Smith et al. проанализировали клиническую картину ПОФ у 28 больных, которых они наблюдали в течение 24 лет.
При этом оссификация скелетных мышц начиналась в первые месяцы или годы от рождения с мышц шеи, верхних отделов спины, мышц, окружающих крупные суставы, с последующим распространением на бедра, плечи; поражались также и жевательные мышцы.
Примечательно, что скорость прогрессирования болезни и выраженность ограничения движений не зависели от возраста дебюта болезни [7].
Гистопатология пораженных тканей при ПОФ хорошо описана в литературе [8–12].
Исследование тканей ранних проявлений ПОФ говорит об интенсивной мононуклеарной и периваскулярной инфильтрации моноцитов, макрофагов, мастоцитов, T- и B-клеток.
Их точная роль в эволюции обострений ПОФ неизвестна, хотя установлено, что любое локальное воспаление независимо от причины его возникновения может спровоцировать активизацию болезни.
Последующая миграция одноядерных воспалительных клеток в пораженную мышечную ткань предшествует масштабной гибели скелетных мышц – процессу, после которого происходит формирование гетеротопического эндохондрального зачатка.
За скоротечной воспалительной стадией следует период интенсивной фибробластной пролиферации, сопровождаемой мощным ангиогенезом.
Фибробластическое поражение на раннем этапе в отношении гистологии неотличимо от агрессивного ювенильного фиброматоза, но по мере созревания ткани фиброзная ткань проходит через фазу бессосудистого уплотнения до состояния хряща, а затем наступает этап реваскуляризации с остеогенезом и характерным процессом эндохондральной оссификации.
Все стадии гистологического развития активных патологий указывают на то, что различные участки пораженного участка ПОФ проходят полный процесс эндохондральной оссификации с разной скоростью. Примечательно, что образующаяся в результате новая гетеротопическая кость гистологически выглядит как нормальная сформировавшаяся кость и часто содержит включения костного мозга.
Мастоциты обнаруживаются на всех этапах формирования тканей при поражении ПОФ, и их количество значительно выше, чем в обычных скелетных мышцах или мышечных тканях больного вне зоны поражения ПОФ. При поражениях ПОФ на этапе фибробластной пролиферации количество мастоцитов многократно превышает показатель, свойственный любой другой форме воспалительной миопатии [13].
При том, что процесс гетеротопного костеобразования в некоторых отношениях схож с формированием костей скелета при эмбриональном развитии или постнатальном заживлении переломов, важнейшим отличием является отсутствие воспалительного компонента при первичном скелетообразовании.
Впрочем, возможно, эти различия вполне условны и объясняются недостаточным объемом накопленных данных о морфологии ПОФ на различных стадиях болезни и малой перспективностью их дополнения, поскольку проведение биопсии пораженных (и даже клинически непораженных) тканей, проведение любого, даже малотравматичного хирургического вмешательства сопряжено с исключительно высоким риском образования новых очагов и быстрого прогрессирования болезни. Хотя процесс гетеротропического костеобразования в некоторых отношениях схож с формированием костей скелета при эмбриональном развитии и послеродовом заживлении переломов, важнейшим отличием является отсутствие воспалительного компонента при первичном скелетообразовании.
Генетика ПОФ и семейные исследования. Семейная ПОФ – чрезвычайно редкое заболевание в популяции и имеет аутосомно-доминантный тип наследования с полной пенетрантностью [14].
Хотя большинство зарегистрированных случаев заболевания являются спорадическими (единичными в семье), в литературе описаны семьи, в которых поражены родитель – потомок/потомки: братья, сестры, в т. ч. близнецы. Представляет интерес наблюдение J.M. Connor et al. [15], описавших семью, где ПОФ была диагностирована в 3-х поколениях у 5 лиц.
В 1-м поколении пораженным был мужчина, который имел 2-х больных дочерей и 2-х больных внучек. Примечательна выраженная фенотипическая изменчивость тяжести течения заболевания в этой семье. Так, мужчина имел асимптоматическое течение до момента травмы, после которой у него развилась тугоподвижность шеи, спины, конечностей и нижней челюсти.
Однако в 70 лет он передвигался, используя трость, и умер в возрасте 72 лет от инфаркта миокарда. Диагноз ПОФ ему был поставлен на основании наличия врожденной деформации большого пальца стопы и рентгенологического обследования, выявившего генерализованный анкилоз позвоночника, множественные зоны оссификации мягких тканей.
Одна из его дочерей также не имела признаков заболевания до 22 лет, когда после экстракции зуба с последующим протезированием у нее развилась тугоподвижность челюсти. Ее сестра в возрасте 15 лет отметила спонтанную припухлость левой голени, когда на основании биопсии была диагностирована фибросаркома.
Однако дальнейшее течение болезни (характерные болезненные припухлости и прогрессирующее ограничение объема движений) свидетельствовало в пользу ПОФ. Летальный исход наступил в 28 лет от пневмонии в состоянии полной обездвиженности. Также имели ПОФ 2 внучки больного от старшей дочери.
F.S. Kaplan et al. [16] представили данные о передаче ПОФ от пораженного отца 2-м дочерям и сыну. Так, 27-летний мужчина (от брака клинически здоровых родителей, 3-й сын в семье, 2 брата здоровы), имеющий характерную микродактилию большого пальца, заболел в 2 года после травмы.
Клинически наблюдалось появление болезненных узлов в области шеи с последующей генерализацией процесса и эктопической оссификацией, прогрессированием болезни и формированием значительного двигательного дефицита. Рентгенологически определялись укорочение и расширение шеек бедренных костей.
У 3-х его потомков также присутствовала врожденная микродактилия больших пальцев с дальнейшим развитием типичной картины ПОФ, при этом у одного из его сыновей первые оссификаты четырехглавой мышцы бедра появилась в 2-месячном возрасте после в/м инъекции, у 2-х других детей – в 3-летнем возрасте.
Как свидетельствуют и другие литературные наблюдения [17], авторами часто описывается фенотипическая гетерогенность ПОФ: характер послеродовой гетеротопической оссификации отличается в зависимости от анамнеза, случаев травматизации мягких тканей и заболеваемости вирусными болезнями. По мнению N. Hebela et al. [18], формирование фенотипа заболевания в период пренатального развития определяется генетическими факторами, в то время как сопутствующие (средовые) факторы влияют на послеродовой ход гетеротопической оссификации.
Источник: https://www.rmj.ru/articles/revmatologiya/Progressiruyuschaya_ossificiruyuschaya_fibrodisplaziya_1/