Узловые гетеротопии серого вещества присутствуют у многих пациентов с другими нарушениями миграции, такими как полимикрогирия или шизэнцефалия. Одиночные или многоочаговые крупные гетеротопические узлы могут являться фокусами парциальных судорог.
Однако даже гигантские гетеротопии, затрагивающие одно полушарие, могут оставаться бессимптомными.
Детализированное нейропсихологическое исследование одного из таких случаев продемонстрировало едва уловимые нарушения полушарных функций, несмотря на нормально развитый интеллект (Calabrese et al., 1994).
Спектр классической лиссэнцефалии и субкортикальной линейной гетеротопии. Эти нарушения миграции могут быть рассмотрены для оценки различных степеней тяжести основной патологии нейрональной миграции, хотя генетически они различаются (Palmini et al., 1993).
Под лиссэнцефалией понимают гладкий мозг. Термин агирия-пахигирия лучше, так как поверхность мозга не всегда гладкая (Aicardi, 1991). В наиболее тяжелых случаях извилины не формируются (агирия). В большинстве случаев присутствует несколько извилин (пахигирия). Dobyns и Leventer (2003) различают 6 степеней лиссэнцефалии (от 1 до 6), в зависимости от количества извилин, видимых на МРТ.
Только степень I заслуживает названия лиссэнцефалии; степени 2-4 являются случаями с пахигирией, и степени 5 и 6 относятся к субкортикальной линейной гетеротопии. В данном разделе объединены различные типы, как имеющие сходный спектр и, очевидно, отчасти сходные механизмы. Хотя имеется несколько форм лиссэнцефалии, в этом разделе рассмотрен только вариант мутации гена LIS1 на 17 хромосоме.
• Классическая (тип 1, Бильшовского) лиссэнцефалия. При классической лиссэнцефалии мозг имеет малые размеры и только первичные, и иногда несколько вторичных извилин. При отсутствии извилин извилистыми становятся сосуды. Кора патологически утолщена (10-20 мм), тогда как белое вещество выглядит узкой полосой вдоль желудочков.
Типично наличие четырех слоев коры: 1) поверхностный, разреженный клеточный слой, аналогичный молекулярному слою нормального мозга; 2) узкий, густоклеточный слой, где располагаются большие пирамидальные нейроны, которые в норме должны располагаться в более глубоких отделах; 3) тонкий слой белого вещества, ниже которого находится
4) широкая полоса малых эктопированных нейронов, распрострающаяся почти до стенки желудочков (Dobyns и Leventer, 2003).
Многие нейроны в клеточных слоях ориентированы неправильно, с апикальным дендритом, направленным вниз или вбок (Takashima et al., 1987).
Более глубокий клеточный слой сформирован из эктопированных нейронов, остановившихся на пути их миграции из герминативного слоя к коре примерно на 12 неделе гестации, поэтому кора выглядит как у 13-недельного плода. Нейроны этого слоя имеют избыточную колонковую организацию.
В продолговатом мозге характерна эктопия ядра оливы. Зубчатые ядра ненормально запутанны, и пирамиды гипоплазированы или отсутствуют (Friede, 1989).
Агенезия мозолистого тела при таком типе необычна. Тип I лиссэнцефалии в 65% случаев возникает в результате мутации гена LIS1, который кодирует 46D белок, некаталитическую часть ацетилгидролазы фактора активации тромбоцитов (Bix и Clark, 1998, Gleeson et al., 1999).
Большинство случаев носит спорадический характер. Зарегистрированы случаи в связи с врожденной цитомегаловирусной инфекцией, но с разными патологическим изменениями (Hayward et al., 1991).
Часть случаев возникает в связи с хромосомной патологией, делецией дистальной части короткого плеча 17 хромосомы (17р13.3).
Некоторые из таких случаев являются частью специфического дисморфического близкого генного синдрома, синдрома Миллера-Дикера, который характеризуется узким лбом, широкой переносицей, отсутствием выемки верхней губы, вздернутыми ноздрями, ретрогнатизмом, аномалиями пальцев и гиперваскуляризацией сетчатки (Dobyns и Leventer, 2003). В таких случаях Dobyns и Truwit (1995) выявили явную делецию 17р13.3 у 14 из 25 пациентов и субмикроскопические делеции в 25 из 38 случаев с использованием цитогенетических методов и в 35 из 38 случаев с флюоресцентной гибридизацией in situ. Сиблинги с синдромом Миллера-Дикера рождались у пар, в которых у одного из родителей произошла пропорциональная транслокация концевого фрагмента хромосомы 17р на хромосому 13-15 пары, что проявлялось в несбалансированных формах у пострадавших детей (Greenberg et al., 1986, Dobyns и Leventer, 2003).
(слева) Тип I (классическая) лиссэнцефалия. Четырехслойная кора. От поверхности (сверху) вниз: (1) молекулярный слой; (2) поверхностный клеточный слой, содержащий несколько типов клеток, включая большие пирамиды, в норме располагающиеся глубже в пятом слое; (3) широкий, бесклеточный слой; (4) широкая полоса гетеротопированных клеток, остановленных при миграции — обратите внимание на столбчатое расположение.
(справа) Нормальное расположение.
Большинство случаев с типом I лиссэнцефалии не являются частью синдрома Миллера-Дикера и определяются как «изолированное» последствие лиссэнцефалии.
Клинические проявления во всех случаях отличаются тяжелой задержкой умственного развития и диплегией, часто атонического типа (de Rijk-van Andel et al., 1990). Как правило, имеются парциальные судороги и, как правило, инфантильные спазмы.
У большинства пациентов присутствует некоторая степень микроцефалии, обычно легкой. При нехромосомной патологии дисморфизм не выражен, хотя лоб узкий и часто присутствует ретрогнатизм. Прогноз неблагоприятный, с ограниченной выживаемостью.
Некоторые случаи мутации LIS1 могут быть в большей степени связаны с субкортикальными групповыми гетеротопиями, нежели чем с лиссэнцефалией (Gleeson et al., 2000).
Диагноз типа I лиссэнцефалии стал возможным при помощи современных методов нейровизуализации. КТ и МРТ демонстрируют характерный внешний вид широкой кортикальной пластинки, с несколькими присутствующими или отсутствующими извилинами, отделенными от гиподенсивного белого вещества слегка волнистой или почти прямолинейной границей.
Слоистость коры может быть выявлена при КТ или МРТ с высокой степенью разрешения. Патологические изменения обычно доминируют в задней части коры, в то время как несколько изгибов можно обнаружить спереди. При ультрасонографии уже с 18,5-25 недели определяется гладкость коры плода или новорожденного (Toi et al., 2004).
МРТ дает более точные результаты (Ghai et al., 2006).
На ЭЭГ в большинстве случаев можно увидеть высокоамплитудную быструю активность альфа и бета частот, чередующихся даже на той же записи с высокоамплитудными дельта или тета медленными ритмами, которые могут имитировать медленные комплексы спайк-волн или гипсаритмию (de Rijk-van Andel et al., 1992, Quirk et al., 1993, Mori et al., 1994).
Дифференциальную диагностику проводят с другими состояниями, при которых имеется утолщение коры и нарушение послойного строения. Пахигирия в результате мутации LIS1 считается лишь легкой степенью лиссэнцефалии, не имеющей отношения к дифференциальному диагнозу.
Определенные нарушения развития плода, особенно цитомегаловирусная инфекция, по-видимому, могут вызывать развитие пахигирии, гистологически связанной с полимикрогирией. Перивентрикулярная кальцификация может сопровождаться патологией формирования извилин мозга.
В таких случаях микроскладки могут сливаться и походить на пахигирию.
Пренатальный диагноз не представляется возможным на поздних сроках беременности с помощью ультрасонографии, поскольку в это время только появляются третичные борозды (Toi et al., 2004). Исследования ДНК могут выявить мутировавший или отсутствующий ген LIS1. Для определения риска рецидива при поиске ламинарных гетеропий необходимы хромосомный анализ и МРТ родителей (особенно матерей).
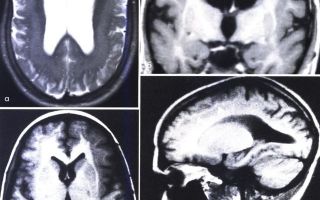
Классическая (тип I) лиссэнцефалия. Т1-взвешенная аксиальная МРТ: (мутация LIS I) толстая корковая лента с гладкой поверхностью и прямой, неволнистой границей между серым и белым веществом. Обратите внимание на присутствие нескольких мелких борозд в лобной области и полное отсутствие борозд сзади,
отсутствие оперкуляции с широко открытой сильвиевой бороздой и слоистость коры со слабой границей между гетеротопированными и полностью мигрировавшими нейронами.
• Субкортикальная ламинарная гетеротопия и лиссэнцефалия в результате мутации DCX гена. Ленточные гетеротопии (Barkovich et al., 1994, Franzoni et al., 1995) или «двойная кора» (Livingston и Aicardi, 1990, Palmini et al.
, 1991) являются результатом нарушенной миграции, при которой поверхностная кора, внешне нормальная или с отклонениями в извилинах, отделена тонким слоем белого вещества от полосы серого вещества. Граница между серым веществом и подлежащим белым веществом ровная как при агирии-пахигирии.
Пациенты с этой аномалией часто страдают судорогами, которые могут иметь очаговый или генерализованный характер, иногда в форме синдрома Ленокса-Гасто и аномальной ЭЭГ (Hashimoto et al., 1993, Parmeggiani et al., 1994).
Нарушения умственного развития значительно варьируют, некоторые пациенты развиваются нормально (Livingston и Aicardi, 1990, Ianetti et al., 1993). Barkovich et al.
(1994) при детальном изучении 27 случаев обнаружили значительную корреляцию между интеллектуальным уровнем и толщиной гетеротопической полосы; внешне нормальная кора была связана с лучшим развитием, но, вероятно, этот признак может варьировать.
На ЭЭГ было обнаружено, что полоса способна продуцировать пароксизмальную активность и повышенный кровоток, как было продемонстрировано с помощью ОФЭКТ, что указывает на активацию коры.
Это состояние в большинстве случаев обусловлено сцепленной с полом мутацией DCX гена, кодирующего даблкортин (англ, doublecortin) (des Portes et al, 1998, Gleeson et al., 1999). Тем не менее, сходная мутация у мальчиков может привести к классической лиссэнцефалии (Pilz et al., 1998).
Мутация очень разнообразно выражается у женщин и даже у некоторых мужчин (Cardoso et al. 2000, Gleeson 2000) и поэтому трудно распознается.
Поэтому генетическое консультирование семьи, где родился мальчик с лиссэнцефалией, должно включать тщательный поиск ламинарной гетеротопии на МРТ и, если необходимо, мутации DCX у матери и сестер.
Известны семьи, где пораженная мать рождала мальчиков с лиссэнцефалией и девочек с ламинарными гетеротопиями (Pinard et al., 1994). Редкие случаи ламинарной гетеротопии связаны с миссенс-мутацией LIS1 и с более умеренным фенотипом (Leventer et al., 2001).
При визуализации, тяжесть экспрессии у девочек варьирует от широких субкортикальных полос, иногда покрытых патологической корой, до с трудом обнаруживаемых тонких полос, которые видны только под ограниченными участками коры.
Односторонние и частичные линейные гетеротопии иногда сложно распознать, для выявления могут потребоваться специальные срезы и изменение формата MPT (Gallucci et al., 1991). У мальчиков картина классической лиссэнцефалии такая же, как при LIS1.
Однако передние отделы коры имеют более гладкую поверхность в сравнении с задними, в отличие от происходящего при мутации LIS1.
Эпилепсия, связанная с ламинарными гетеротопиями, может поддаваться медикаментозному лечению, но бывает и устойчивой. Хирургическое лечение оказалось неэффективным.
Синдром Барайтсера-Уинтера включает дисмор-фические признаки и пороки развития мозга в виде классической лиссэнцефалии или субкортикальных ламинарных гетеротопий (Rossi et al., 2003).
• Пахигирия. Этот тип представляет менее тяжелую форму спектра лиссэнцефалии и, вероятно, возникает в результате тех же механизмов. Однако эта форма гетерогенна и может входить в состав разных синдромов. Клинически пахигирия представлена различными схожими симптомами, но с меньшей тяжестью. На МРТ выявляют утолщение коры и линейное разделение между корой и белым веществом.
(а) Лиссэнцефалия-пахигирия у двухлетней девочки: ЭЭГ указывает на типичные быстрые ритмы с альфа и выше частотой. (б| Синдром Миллера-Дикера у 14-недельной девочки: ритмическая активность различных частот, но в основном в тета-диапазоне.
(в) Синдром Миллера-Дикера у двухлетнего мальчика: хотя присутствует некоторая избыточная тета-альфа активность, в записи преобладают повторяющиеся вспышки острых волн, достигающих 500-600 μВт.
• Другие формы и синдромы лиссэнцефалии. Распознание некоторых менее распространенных вариантов лиссэнцефалии не менее важно из-за разницы генетических и прогностических последствий (Hennekam и Barth, 2003, Raoul et al., 2003).
Микролиссэнцефалия состоит из крайне выраженной врожденной микроцефалии и агирии или пахигирии с широкой корой.
Описано по меньшей мере, пять или шесть типов, передающихся по рецессивному типу, с различной степенью утолщения кортикального слоя, расположением имеющихся борозд и наличием сопутствующих пороков, таких как гипоплазия мозжечка, стволовая атрофия и увеличение желудочков (Ross et al., 2002, Dobyns и Leventer, 2003, Sztriha et al., 2004).
Некоторые авторы (Dobyns и Barkovich, 1999) выделили эти случаи из «олигирической микроцефалии» (Hanefeld, 1999), которую они расценивают скорее как форму первичной микроцефалии, нежели форму расстройства миграции. Один из этих синдромов может быть связан с мутацией рилин гена (Hong et al., 2000, Crino, 2001).
Лиссэнцефалия с гипоплазией мозжечка является отдаленным проявлением микроцефалии с рудиментарной двуслойной корой мозга и тяжелой гипоплазией мозжечка (Ross et al., 2001, Sztriah et al., 2005). Вероятно, с рецессивным наследованием.
Лиссэнцефалия с гипоплазией мозолистого тела генетически гетерогенна. Некоторые случаи могут входить в группу мутации LIS1 или микролиссэнцефалии.
Х-сцепленная лиссэнцефалия с аномалией гениталий (XLAG) — врожденный порок с микроцефалией, тяжелой задержкой развития, тенденцией к гипотермии, отсутствием мозолистого тела и множественными аномалиями мозга (Berry-Kravis и Israel, 1994, Dobyns et al., 1999). Более вероятна гипоплазия гениталий, чем агенезия.
XLAG развивается в результате мутации гомеобоксного гена ARX на хромосоме Х33.2 (Uyanik et al., 2003), на которой другие мутации также могут быть причиной некоторых неврологических синдромов (Kato et al.
2004, Suri 2005), включая Х-сцепленную задержку умственного развития (MRX54), агенезию мозолистого тела с патологией гениталий и синдром Партингтона с умственной отсталостью, атаксией и дистонией в зависимости от типа мутации.
Интересно, что лиссэнцефалия с неонатальными судорогами и тяжелыми аномалиями развития нервной системы, как было выявлено, связана с отсутствием глутамина.
Субкортикальная групповая гетеротопия («двойная кора»): (а) Аксиальный срез МРТ: широкие, непрерывные группы с таким же сигналом как от коры. (б) Коронарный срез: в этом же случае имеется расширение желудочков преимущественно спереди.
(в, г) MPT, Т1-взвешенная последовательность — (в) аксиальный срез, (г) сагиттальный срез -тонкий слой белого вещества, лежащий между истинной корой и тонкой линейной гетеротопией серого вещества (стрелки).
— Также рекомендуем «Комплекс cobblestone: лиссэнцефалия типа 2″
Редактор: Искандер Милевски. 29.11.2018
Оглавление темы «Аномалии развития коры головного мозга.»:
Лиссэнцефалия
Лиссэнцефалия – группа генетически обусловленных аномалий развития головного мозга, характеризующихся частичным или полным недоразвитием извилин и борозд коры больших полушарий, а также нарушением ее ультраструктуры. Выраженность и сочетания симптомов этого состояния различаются при разных формах заболевания, наиболее распространены судороги, глубокая умственная отсталость, нарушения глотания и гипотония мышц. Диагностика лиссэнцефалии может производиться ультразвуковыми методиками (в том числе и пренатально), компьютерной и магнитно-резонансной томографией, для наиболее распространенных форм возможно определение посредством молекулярно-генетического анализа. Специфического лечения не существует, используют симптоматическую и поддерживающую терапию.
Лиссэнцефалия – группа тяжелых аномалий развития головного мозга, которые сопровождаются недоразвитием коры больших полушарий с формированием пахигирии (наличием всего нескольких извилин и борозд) или агирии (полным отсутствием складчатости коры).
Данная патология может выступать в качестве самостоятельного генетического заболевания или входить в симптомокомплекс других синдромов – например, Миллера-Дикера, Фукуямы и Уокера-Варбурга.
Механизм наследования различных типов лиссэнцефалии может быть аутосомно-рецессивным, аутосомно-доминантным (в данном случае чаще всего имеют место спонтанные или герминативные мутации) и сцепленным с Х-хромосомой.
Из-за многообразия механизмов наследования половое распределение нарушения неодинаково при различных формах патологии.
Лиссэнцефалия является достаточно редкой генетической аномалией развития мозга, поэтому встречаемость определена только для наиболее распространенной первой группы нарушений – она составляет 11,7 случаев на 1 000 000 новорожденных. Для остальных групп лиссэнцефалии встречаемость не установлена, в том числе и потому, что во многих случаях плод с подобной патологией не вынашивается, и беременность самопроизвольно прерывается в первом триместре еще до определения наличия порока.
Лиссэнцефалия
Основная общая причина всех типов лиссэнцефалии – нарушение процесса миграции клеток-предшественников нейронов (нейробластов) из передних отделов нервной трубки к будущей коре больших полушарий.
В результате этого вместо сложной складчатой структуры, которая имеет в своем составе шесть слоев, образуется гладкая или имеющая в разы меньше борозд кора, состоящая из 2-4 слоев (в зависимости от формы заболевания).
Поскольку кора больших полушарий у человека отвечает за когнитивные функции, в ней содержится огромное число нервных центров и обширные ассоциативные зоны, лиссэнцефалия приводит к тяжелейшим расстройствам. Кроме того, при некоторых типах мутаций, вызывающих данное состояние, возможно развитие аномалий других органов и тканей, что еще больше усугубляет состояние больного.
Наиболее распространенные типы лиссэнцефалии обусловлены дефектами гена PAFAH1B1, также известного под названием LIS1 и расположенного на 17-й хромосоме. Многие врачи-генетики отмечают, что для возникновения выраженного недоразвития коры головного мозга и синдрома Миллера-Дикера необходимы не точечные мутации в LIS1, а крупные делеции с захватом сотен пар азотистых оснований.
Нередко при этом повреждаются и окружающие гены, что становится причиной разнообразных фенотипических проявлений данного типа лиссэнцефалии. Ген LIS1 кодирует внутриклеточную субъединицу сложного по своей структуре фермента, который принимает активное участие в миграции нейробластов.
Дефекты в структуре этой субъединицы приводят к нарушению данного процесса и развитию данного заболевания.
Другая форма лиссэнцефалии обусловлена мутацией гена DCX, локализованного на Х-хромосоме, поэтому наследование этого типа патологии сцеплено с полом.
Белок, получаемый в результате экспрессии данного гена, участвует в формировании особого типа микротрубочек, которые вырабатываются только нейробластами и необходимы им для формирования связей между клетками.
Нарушения в структуре DCX приводят к синтезу дефектного белка, что провоцирует лиссэнцефалию. Другой относительно изученный вариант этого состояния, также сцепленный с Х-хромосомой, вызывается мутациями гена ARX, который является фактором транскрипции для иных генов.
Он контролирует развитие коры больших полушарий, поджелудочной железы и половых органов, из-за чего дефекты ARX проявляются многочисленными пороками данных органов, в том числе лиссэнцефалией.
Еще одним распространенным вариантом лиссэнцефалии является форма заболевания, обусловленная мутацией гена RELN, расположенного на 7-й хромосоме.
Дефекты этого гена приводят к так называемому синдрому Норман-Робертса, который, помимо прочего, сопровождается выраженным нарушением складчатости коры больших полушарий.
Ген RELN кодирует последовательность гликопротеида рилина, который принимает активное участие в формировании нервной ткани, а также контролирует образование новых дендритов и функционирование долговременной памяти у взрослых людей.
Удалось идентифицировать еще один ген, приводящий к развитию лиссэнцефалии – TUBA1A, который локализован на 12-й хромосоме. Он кодирует определенный компонент цитоскелета нейронов, его дефект приводит к неполноценности нейробластов, что нарушает процесс их миграции в эмбриональном периоде.
На сегодняшний день выявлено более двадцати вариантов лиссэнцефалии. Эти варианты обусловлены различными мутациями, имеют отличия в фенотипических проявлениях заболевания, разные механизмы наследования и тяжесть симптомов.
Долгое время разновидности патологии не удавалось успешно классифицировать из-за значительного разнообразия генетических дефектов и недостаточных данных о ключевых генах, приводящих к развитию некоторых форм заболевания.
Лишь в 2003-м году удалось создать достаточно приемлемую с точки зрения современной генетики и неврологии классификацию лиссэнцефалии, которая учитывает основные нюансы этого состояния. Специалисты разделяют все формы лиссэнцефалии на пять классов:
- Класс 1, часто называемый классической лиссэнцефалией. Включает в себя формы заболевания, обусловленные мутациями гена LIS1 (изолированный тип и синдром Миллера-Дикера), а также сцепленную с полом разновидность, вызванную мутацией DCX. Кроме того, в этот класс часто включают некоторые типы лиссэнцефалии с неясными генетическими причинами. Особенностью группы является нарушение складчатости коры больших полушарий с минимальным проявлением иных аномалий центральной нервной системы.
- Класс 2 – в настоящее время состоит только из одного типа лиссэнцефалии, которая вызывается дефектами гена ARX. Заболевание сцеплено с Х-хромосомой, помимо пороков развития коры у больных часто обнаруживаются отсутствие мозолистого тела, нарушения терморегуляции, выраженные аномалии половых органов и поджелудочной железы.
- Класс 3 – включает в себя разновидности лиссэнцефалии в сочетании с гипоплазией или полным недоразвитием мозжечка, наиболее типичной мутацией для данного класса являются дефекты гена RELN. Может регистрироваться как изолированный вариант патологии или в составе синдрома Норман-Робертса.
- Класс 4, нередко называется микролиссэнцефалией, поскольку нарушение формирования коры больших полушарий сопровождается выраженной микроцефалией. Некоторые формы этого заболевания были достоверно ассоциированы с мутациями гена TUBA1A.
- Класс 5 или булыжниковые лиссэнцефалии, к которым относят синдромы Фукуямы и Уокера-Варбурга. Последний соотносится с дефектами генов POMT1, POMT2, FKRP и некоторыми другими, все они расположены на 9-й хромосоме.
Данная классификация критикуется некоторыми исследователями по причине наличия большого количества «белых пятен» в виде включения в нее форм лиссэнцефалии с неопределенными ключевыми генами. Однако в настоящее время именно это разделение считается наиболее общепринятым в научном мире. Классификация корректируется по мере дальнейшего изучения данного состояния.
Основными проявлениями лиссэнцефалии являются мышечная слабость, выявляемая уже при рождении, частые расстройства глотания и сосания, судорожные припадки, сильное отставание в физическом и умственном развитии.
Выраженность тех или иных проявлений заболевания зависит от наличия пороков развития как головного мозга, так и других органов и систем, а также от степени недоразвития коры. Наиболее тяжелые формы патологии наблюдаются при наличии полной агирии.
Иногда больные лиссэнцефалией могут доживать до подросткового и даже взрослого возраста, глубокое недоразвитие центральной нервной системы сохраняется в течение всей жизни. Некоторые пациенты проживают жизнь на грани вегетативного состояния.
Летальный исход при лиссэнцефалии наступает из-за осложнений, обусловленных пороками других органов и систем, а также из-за вторичных пневмоний, сердечно-сосудистых и иных аномалий.
В отношении лиссэнцефалии возможна пренатальная диагностика при помощи ультразвуковых методов исследования, при этом увеличение разрешающей способности УЗИ-аппаратуры способствует все более раннему определению заболевания.
Развитие нарушений в строении коры больших полушарий происходит на 14-20-й неделе гестации, в настоящее время уже в этот период можно определить патологию и поставить вопрос о прерывании беременности. После рождения ребенка с подозрением на лиссэнцефалию диагноз подтверждают при помощи КТ и МРТ.
Молекулярно-генетическая диагностика обладает высокой точностью, но она доступна только в отношении тех форм заболевания, для которых известны ключевые гены.
Специфического лечения лиссэнцефалии не существует, применяют противосудорожные препараты, ноотропы и другие средства, позволяющие уменьшить выраженность симптоматики. При наличии иных пороков развития производится их коррекция по медицинским показаниям.
Прогноз и профилактика лиссэнцефалии
Прогноз практически любой формы лиссэнцефалии крайне неблагоприятный, большинство больных умирает в раннем детстве от осложнений и других пороков развития.
Описаны отдельные легкие случаи этого состояния с частичным или очаговым недоразвитием коры больших полушарий, но не все специалисты склонны причислять такие типы патологии к лиссэнцефалиям. Профилактика возможна только в рамках пренатальной диагностики.
При наличии в роду подобных заболеваний имеет смысл провести генетический анализ на носительство генов аутосомно-рецессивных типов патологии. При обнаружении у плода нарушений формирования мозга, соответствующих лиссэнцефалии, ставится вопрос о прерывании беременности.
Лиссэнцефалия — есть ли шанс, если у ребенка «гладкий» мозг?
В итоге поверхность мозга становится удивительно гладкой. Этот дефект начинает проявляться еще с 12 недели развития плода, и заканчивает формироваться спустя 2 недели, и вызван он нарушенным процессом передвижения нейронов в этот период.
Головной мозг, в нормальном состоянии у каждого здорового человека, имеет множество складок и борозд. У плода с этим отклонением, структура развивается не своеобразным способом, у таких пациентов складки и борозды либо частично отсутствуют, либо их вовсе нет, и произошедшие изменения уже не обратимы.
Точно так же, как и многие другие дефекты в развитии головного мозга, лиссэнцефалия берет под контроль большой спектр фенотипов, они могут колебаться в соответствии с тяжестью от переменной агирии, либо до полной.
Сопутствующие заболевания и нарушения
Сопутствовать данному пороку развития может — синдром Миллера-Дикера, либо Уокера-Варбурга.
Синдром Миллера-Дикера характеризуется классической лиссэнцефалией, при таком синдроме под воздействие аномалии попадают лицевые
Синдром Уокера-Варбурга
мышцы и проявляются другие возможные дефекты, которые можно встретить у людей только с таким синдромом.
Особенностью синдрома является потеря в хромосоме 17 нескольких генов. За лиссэнцефалию отвечает потеря гена PAFAH1B1, это доказанный учеными факт.
А вот при утере гена YWHAE, лиссэнцефалия может усложниться. Последствия после потери других генов при данном синдроме еще не известны.
Синдром Уокера-Варбурга – очень редкая врожденная дистрофия мышц, связанная с нарушением работы головного мозга и лицевых мышц.
Классификация заболевания
На данный момент существует большое количество систем для классификации патологии. Рассмотрим наиболее популярную в нынешних реалиях, в которой имеется, — тип первый – классический, и тип второй – «булыжник».
Классический тип мутации
Развитие патологии происходит из-за мутировавшего гена PAFAH1B1. Такой дефект можно разделить на изолированную лиссэнцефалию и синдром Миллера-Дикера.
Изолированная патология проходит без развития других дефектов в геноме, во втором случае наблюдается развитие сопутствующих аномалий из-за мутации в DCX гене.
Тип «булыжник»
Второй тип «булыжник» включает следующие типа аномалии:
- заболевание – мышцы-глаз-мозг;
- синдромы Уокера-Варбурга и Фукуямы.
Кроме этого, существуют также другие подтипы:
- микролиссэнцефалия;
- синдром Нормара-Робертса;
- патология вызванная мутировавшим ARX геномом.
Типы развития
Рассмотрим патологию, базируясь на 4 типах развития.
Тип первый
Мозг ребенка можно сравнить с мозгом плода на 23 неделе развития. Кора состоит из четырех слоев нейронов, развивается пахигирия, агирия.
Особенности:
- гладкая наружная часть коры мозга;
- сильно уменьшен объем белого вещества в мозгу;
- ленточная гетеротопия, которая отделена от коры белой полосой;
- гипоплазия ствола.
Тип второй
Особенности:
- гладкая поверхность коры;
- гипоплазия;
- размыта граница между белым и серым веществом;
- утолщение коры;
- агенезия мозолистого тела;
- у некоторых больных в затылочной области – энцефалоцеле.
Тип третий
Особенности:
- белое вещество в меньших количествах, нежели нужно;
- гипоплазия;
- начинают выделяться извилины, но это малозаметно.
Тип четвертый
Особенности:
- остановка миелинизации;
- головной мозг очень мал в размере;
- количество нейронов составляет 35% нормы;
- на коре виднеются некоторые извилины и борозды с нормальной толщиной.
В некоторых случаях используют еще одну классификацию лиссэнцефалии, ее основывают в зависимости от тяжести болезни:
- самый тяжелый класс, тотальная агирия, нет никаких извилин;
- имеется небольшое количество складок на затылочных и лобных полосках, диффузная агирия;
- сочетание пахигирии и агирии;
- диффузная пахигирия;
- пахигирия спереди и гетеротопия;
- гетеротопия под корой.
Первая и четвертая степень очень редка, вторая имеется у детей с синдромом Миллера-Дикера. Самое частое — сочетание в третьей степени, обычно такая патология состоит из задней агирии и лобной пахигирии.
Тип первый, то есть классический, встречается у 12 человек на миллион рожденных.
Видео по теме:
Причины и патогенез аномального развития
Еще у примерно 3% людей, конкретно с классическим типом патологии, можно отметить мутацию в гене TUBA1A. Примерно у 30% пациентов с гипоплазией наблюдаются изменения в TUBA1A гене.
Иные причины возникновения могут быть самыми разными, например, при развитии инфекционных заболеваний в матке в первом триместре беременности, или же на ранних сроках у плода было плохое кровоснабжение в области мозга, в результате чего начала развиваться лиссэнцефалия.
Процесс развития коры головного мозга:
- нервные клетки начинают делиться;
- кора дифференцируется;
- нейроны движутся из матрикса.
Хронология развития плода в норме должна происходить таким образом:
- на 11-12 неделях начинают формироваться оба полушария;
- на 16 – начинают формироваться борозды и извилины;
- на 20 — уплотняется кортикальное плато, начинают формироваться первичные и центральные борозды;
- на 23-26 — сформировываются ольфакторные борозды;
- на 28 неделе развития внутри утробы матери, начинают формирование борозды в височной области.
Кора головного мозга формируется за счет миграции мозговых клеток – нейронов, которые находятся в разных слоях мозга. В процессе их передвижения, клетки, которые сформировались на более позднем этапе, передвигаются дальше, чем предыдущие, в результате происходит формирование нового слоя. Этот процессе регулируется специальными белками N-кадгерином и рилоном.
В норме, все основные борозды и извилины начинают свое формирование после окончания передвижения нейронов, именно нарушение данного процесса влияет на неправильное создание извилин и борозд.
Постановка диагноза и возможности медицины
Обычно в таком случае диагноз ставится при рождении или немного позже, после проведения УЗИ и исследования результатов КТ или МРТ.
Если врач заподозрит лиссэнцефалию, кроме УЗИ нужно будет проводить еще несколько методов диагностики, например, анализ на явность мутирующих генов и ЯРМ-томографию.
Обнаружить данную патологию можно не раньше, чем на 20 неделе беременности, поскольку поверхность мозга до этого времени, и без каких-либо нарушений, является гладкой. Именно после данного периода, можно заметить отклонения в развитии плода.
Вылечить данный порок невозможно, есть возможность симптоматического лечения, и оно зависит от стадии развития и места расположения дефектов. Необходимо поддерживать постоянный уход.
Для людей с гидроцефалией обычно делают шунтирование, а для контроля судорог принимаются специальные лекарственные препараты.
Прогноз продолжительности жизни
Главным фактором, который определяет прогноз продолжительности жизни ребенка с диагнозом лиссэнцефалия, является степень тяжести заболевания. Так некоторые из пациентов имеют более-менее нормальное интеллектуальное развитие.
В основном дети, с более тяжелой формой патологии, не способны прожить дольше 10 лет, но даже дожив до этого возраста, их развитие все равно остается на уровне 4-6 месячного ребенка.
Главной причиной смерти больных, в основном, является связь с аспирацией питательных продуктов или жидкости, с очень тяжелыми приступами, или с болезнями дыхательных путей.
лиссэнцефалия
Сглаживание извилин коры головного мозга (агирия) формируется внутриутробно. Патогенетическим механизмом развития нозологии является нарушение эмбриогенеза, обусловленного патологией распространения из первичной нервной трубки нейробластов. Сглаживание поверхности сопровождается снижением умственной, интеллектуальной активности.
Сопутствующие неврологические расстройства постепенно нарастают. Заболевание приводит к летальному исходу еще в грудном возрасте.
Отсутствие мозговых извилин – исключает оптимальное функционирование второй сигнальной системы, которая существует только у человека. Складчатость повышает функциональную поверхность мозга.
Даже раннее выявление с помощью МРТ головного мозга не позволяет спасти жизнь ребенку с лиссэнцефалией первого или второго типа.
На протяжении беременности врожденные пороки развития церебральных структур определяются ультразвуковым обследованием (УЗИ). Достоверность процедуры – 60%, поэтому иногда после МРТ выявляются врожденные пороки развития мозга, хотя внутриутробный мониторинг патологии не определяет патологических состояний.
Пример заключения магнитно-резонансной томографии:
- Билатеральная вентрикуломегалия;
- Увеличение ликворных пространств;
- Сглаженность церебральных извилин;
- Неполное отсутствие складок теменных и лобно-височных областей;
- Расширение боковых желудочков.
Нозология нередко сочетается со стигмами церебрального дисэмбриогенеза – макроцефалией, агирией, микроцефалией.
Медицинские источники утверждают, что патологию может обнаружить УЗИ после двадцать шестой недели беременности.
МРТ: норма и лиссэнцефалия
Лиссенцефалия 2 типа имеет альтернативное название «синдром Норман-Робертс». Канадские ученые, открывшие заболевание, считают основной причиной формирования заболевания мутацию гена «RELN».
Участок ДНК, отвечающий за нозологию, кодирует образование протеина «рилина». Синдром характеризуется множеством клинических изменений:
- Лимфедема (лимфатический отек);
- Эпилептические приступы;
- Покатость лба;
- Утолщение церебральной коры;
- Агирия (сглаженность мозговых борозд);
- Макроцефалия;
- Микроцефалия.
Перинатальное УЗИ обнаруживает большую часть случаев болезни.
Причины возникновения лиссенцефалии
Формирование внутримозговой коры у плода проходит несколько стадий развития. Вначале происходит деление нейронов. Постепенно нервные клетки мигрируют из первоначального матрикса в места постоянного расположения. Остальные этапы эмбриогенеза сопровождаются специализацией клеточных пластов, отвечающих за анатомические структуры.
Сложный процесс образования мозга должен проходить под строгим контролем биохимических механизмов. Любое внешнее вмешательство нарушает внутриутробное развитие.
Основные причины возникновения лиссэнцефалии:
- Внутриутробная гипоксия (кислородное голодание);
- Вирусные инфекции;
- Хромосомные аномалии (дефекты формирования протеина рилина в 7-ой хромосоме).
Внутрисемейные случаи передачи заболевания достаточно редки. Если встречается нозология у нескольких членов семьи, требуется генетическое консультирование.
Средняя распространенность нозологии первого типа – 11% на миллион новорожденных детей. Большинство ситуаций провоцируется генетическими дефектами гена LIS1. На втором месте встречается гипоплазия мозжечка из-за дефекта гена TUBA1A.
После наследственных аномалий на втором месте – вирусное инфицирование в первом триместре беременности.
Основные виды лиссенцефалии
Встречается свыше двадцати морфологических форм заболевания:
- Классическая форма (I типа) с мутацией LIS1, синдромом Миллера-Дикера;
- Форма с наследственным дефектом DCX;
- Изолированная разновидность без мутаций;
- Агенезия мозолистого тела при аномалии Х-хромосомы;
- Гипоплазия мозжечка с нарушением кодирования рилина (синдром Нормана-Робертса);
- Микролиссэнцефалия;
- Булыжниковый вид с синдромом Фукуямы, Уокера-Варбурга, мышечно-глазо-мозговой формой.
Мышечная дистрофия с олигофренией Фукуяма передается аутосомно-рецессивным способом. Наследственная миотония имеет прогрессирующее течение.
Особенности шизэнцефалии, порэнцефалии, голопрозэнцефалии
Билатеральная расщелина церебральных полушарий формируется на этапе эмбриогенеза. Морфологическая структура образования бывает разомкнутой или сомкнутой.
Односторонняя локализация патологии на МРТ снимках может напоминать кисту. Вдоль расщелины расположены патологические участки церебральной паренхимы – микрогирия, зоны повышенной повторной активности, спастические параличи.
Порэнцефалия характеризуется образованием кистозных полостей внутри церебральной паренхимы. Причины возникновения патологии – кровоизлияние внутрь головного мозга, инфаркты, инсульты. Типичная локализация кисты – сильвиева борозда.
В редких случаях порэнцефалия характеризуется образованием аномальных ходов между кистозной полостью и желудочковыми пространствами. Нозология нередко характеризуется другими стигмами дисэмбриогенеза:
- Микроцефалия;
- Энцефалоцеле;
- Агирия.
У ребенка с пороком развития возникают неврологические расстройства:
- Олигофрения (умственная отсталость);
- Приступы эпилепсии;
- Атрофические изменения дисков зрительного нерва;
- Тетрапарез.
Осложняют клиническую картину сопутствующие изменения:
- Артериальные и венозные кровоизлияния;
- Внутричерепная гипертензия (при нарушении циркуляции ликвора);
- Церебральные инфаркты;
- Гемипарезы;
- Очаговые эпилептические очаги.
Псевдопорэнцефалические кисты нередко имеют унилатеральное (одностороннее) расположение. Нередко сочетаются с пороками центральной нервной системы, дефектами клеточной миграции.
Шизэнцефалия на МРТ
Голопрозэнцефалия – что это такое
Заболевание возникает по причине патологического разделения химического вещества прозэнцефалона.
Классификация нозологии по степени тяжести:
- Лобарная;
- Полулобарная;
- Алобарная.
Самая тяжелая – последняя форма. При ней наблюдается ряд врожденных аномалий:
- Недоразвитие межчелюстной кости;
- Синофтальм;
- Цебоцефалия.
На фоне множественных аномалий части лица трудно различимы. Сопутствующие пороки нейрональной миграции:
- Синостоз (сращение нервных ганглиев);
- Наличие одного церебрального желудочка.
Этиологические механизмы наследственных дефектов не установлены. Голопрозэнцефалия в большинстве случаев завершается смертельным исходом, но неполная форма может приводить к инвалидности.
Симптомы лиссэнцефалии
Клинические проявления нозологии определяются сразу после рождения ребенка. Отмечаются не только генетические дефекты, но и нарушения глотания у малыша, повышенный мышечный тонус, отсталость в развитии от сверстников.
Симптомы лиссэнцефалии у ребенка 2-5 месяцев:
- Олигофрения;
- Нарушение умственного развития;
- Миоклония;
- Увеличение артериального давления.
Вначале умственная отсталость не прослеживается. После первого года жизни наблюдаются расстройства психоречевых навыков. Дети раздражительны, тревожны, мышечная активность снижена.
При сравнении со сверстниками наблюдается значительное отставание в развитии, формировании мышечной системы, иннервации внутренних органов, малого таза.
При легком течении заболевания ребенок может дожить до подросткового возраста. Внешние аномалии ограничивают социального общения. Больные редко доживают до восемнадцати лет, но существует около двадцати разновидностей патологии. Проявления первого типа:
- Макрогирия;
- Шизэнцефалия;
- Микроцефалия;
- Четырехслойное строение коры;
- Гипоплазия моста;
- Недоразвитие мозжечка.
Определить лиссэнцефалию при отсутствии положительных результатов внутриутробного УЗИ сложно. Клиническая картина возникает после рождения. Мышечные параличи, судороги появляются сразу или на протяжении первого года.
В большинстве случаев установить диагноз лиссэнцефалии поможет ультразвуковое обследование. Врач может назначить магнитно-резонансную томографию беременной женщине и ребенку. МРТ является безвредной, но в первом триместре не проводится из-за отсутствия практической информации о влиянии магнитного поля на плод.
Первые признаки лиссэнцефалии у новорожденных
Раннее выявление нозологии помогает продлить жизнь малышу, но спасти от смерти не удается. Первые признаки у новорожденных:
- Небольшая голова;
- Апатия, вялость;
- Широкое межглазное расстояние;
- Эпилептический судорожный синдром;
- Гипертонус мускулатуры;
- Увеличение ширины между легкими и почками;
- Активация патологических рефлексов;
- Покатая лобная часть.
На протяжении первого года жизни присоединяются другие проявления – речевые расстройства, недержание мочи, учащение дыхания и сердцебиения.
Синдром летального птеригума (Нормана-Робертса) возникает при лиссэнцефалии первого типа. К вышеописанным проявлениям присоединяется ряд проявлений:
- Сложности сидения;
- Невозможность удержания малышом вертикальной позиции;
- Появление генерализованных подергиваний мускулатуры;
- Аномалии краниофациальной области (широкие глаза, покатый лоб, бугры на затылке);
- Глазной нистагм;
- Снижение мышечного тонуса.
Мозжечковая атаксия характеризуется нарушением координации. Ребенок не может удержать равновесия, поддерживать вертикальную позицию. Родители вначале обращают внимание на сложности посадки, сохранения горизонтального положения.
Диагностика лиссэнцефалии
Устанавливается диагноз сразу после рождения с помощью УЗИ, КТ и МРТ. Внутриутробно обнаружить проявления можно с 20 недели.
Выявление малейших нарушений развития головного мозга плода требует дополнительной диагностики. В зависимости от степени повреждений принимается решение о возможности прерывания беременности.
При подозрении на наследственные формы проводится генетическое консультирование всех членов семьи.
Поздняя верификация патологии требуется постоянного консервативного лечения. Терапия тяжелых форм проводится в стационаре под контролем квалифицированных врачей.
Основные виды диагностики лиссэнцефалии:
- Магнитно-резонансная томография (МРТ головы) показывает мягкие ткани. Исследование обнаруживает церебральные аномалии, кисты, скопления жидкости, воспалительные очаги;
- Внутриутробное УЗИ головы выполняется на 22-27 неделе, когда прослеживается процесс образования борозд;
- Компьютерная томография (КТ) помогает верифицировать изменения серого и белого вещества, дополнительные твердые образования. Обследование приводит к радиационному облучению тканей, поэтому детям выполняется по строгим показаниям;
- Электроэнцефалография (ЭЭГ) верифицирует очаги повышенной мозговой активности, участки гиперактивности паренхимы.
Самое достоверное исследование – МРТ при лиссэнцефалии у новорожденных, позволяющее определить самые мельчайшие аномалии.
Дифференциальная диагностика
Схожими клиническими признаками характеризуется ряд хромосомных аномалий – синдром Пена-Шокейра, Нунана, артрогрипоз. Генетическое консультирование верифицирует нозологии до появления клинических симптомов.
Контрактуры суставов удается диагностировать ближе к первому году, когда ребенок предпринимает попытки сидеть или ходить. Внешние проявления водянки мозги – увеличенная форма черепа, выпячивание родничков.
Прогноз и продолжительность жизни
Большинство детей не доживает до года. Если ребенку удается дожить до 3 лет, он остается инвалидом и погибает от сопутствующих инфекций, тяжелых осложнений – парезы кишечника, мочевого пузыря, конечностей, дыхательной мускулатуры.
Угнетение глотательного рефлекса обуславливает затруднения питания, что обеспечивает дефицит массы тела, приводит к снижению иммунитета.
Прогноз неблагоприятный даже при внутриутробном выявлении аномалий развития.